
Osteogenesis Imperfecta (OI) is a genetic disorder that predominantly impacts bone formation, resulting in fragile skeletal structures and numerous associated complications. Characterized by a wide spectrum of clinical manifestations, OI presents a unique case study in medical genetics that combines elements of bone biology, genetic heterogeneity, phenotypic variability, and the impacts of environmental factors.
Osteogenesis Imperfecta: A historical perspective
The origins of Osteogenesis Imperfecta (OI) date back to ancient Egypt, as displayed by a skeleton exhibited in the British Museum. More contemporary knowledge about the disease is well documented. Malebranche documented the first case of OI, reporting the clinical heterogeneity of this condition.

Synthesis of collagen and its significance in osteogenesis imperfecta
Collagen, the most abundant protein in the human body, provides structural support and strength to various tissues such as the skin, tendons, bones, and ligaments. Its complex synthesis involves several steps, each orchestrated by a series of enzymes and biochemical processes.
The journey to a mature collagen fiber begins with the translation of specific types of collagen genes into preprocollagen chains in the ribosomes of the cell. These genes encode for alpha chains that have a characteristic repeating sequence of three amino acids, glycine-X-Y, where X and Y are often proline and hydroxyproline.
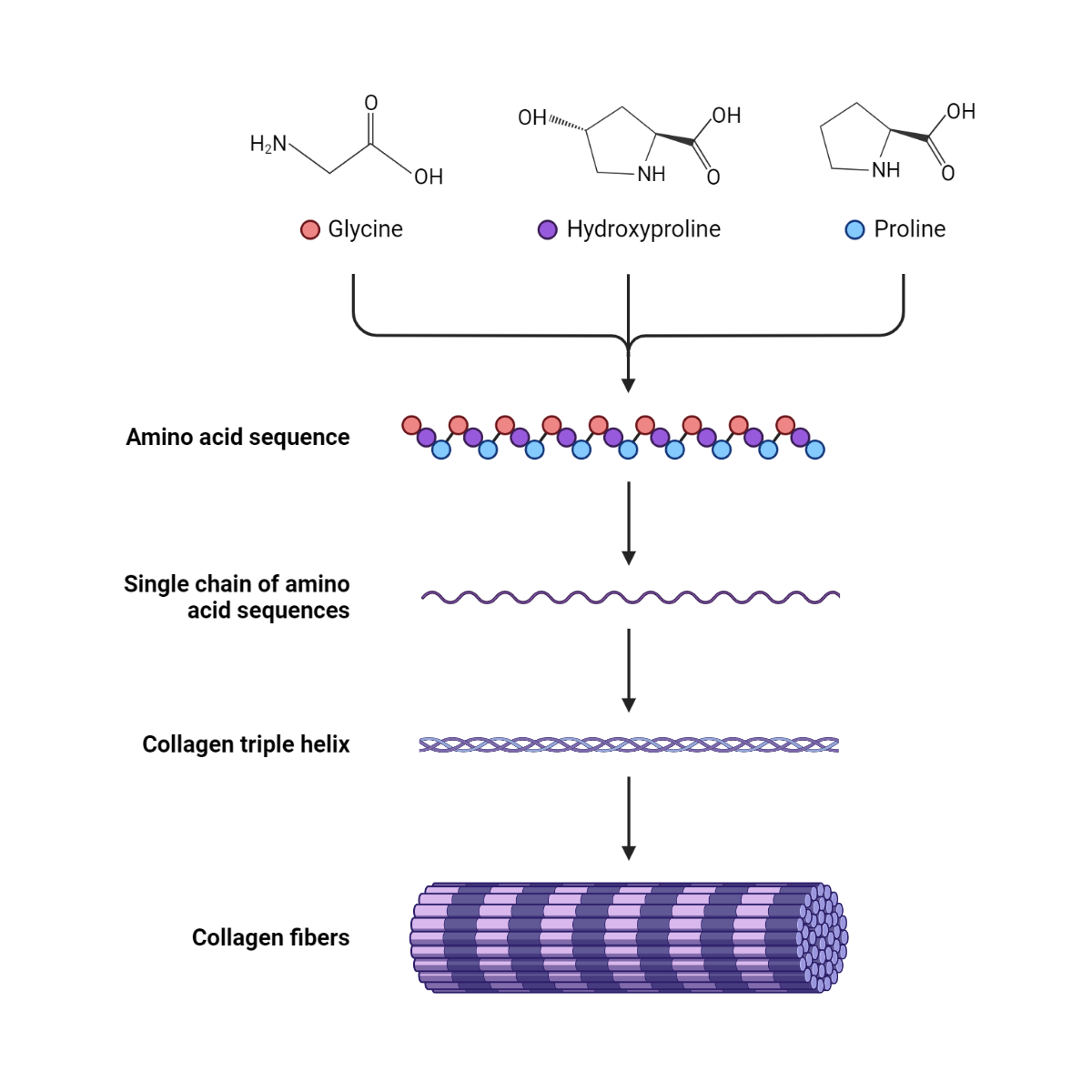
Once the polypeptide chains are synthesized, they enter the lumen of the endoplasmic reticulum (ER), where several posttranslational modifications occur. These include the hydroxylation of specific proline and lysine residues, a process catalyzed by prolyl hydroxylase and lysyl hydroxylase. This hydroxylation is critical for the stability of the final collagen triple helix. Vitamin C is an essential cofactor for these hydroxylation reactions, which is why a deficiency can lead to scurvy, a disease characterized by a defect of collagen synthesis.
Next, glycosylation of the hydroxylysine residues takes place, forming galactosyl-hydroxylysine or glucosyl-galactosyl-hydroxylysine. This glycosylation aids in the formation of the triple helix.
Following these modifications, three alpha chains align and coil around each other, forming a triple helix known as procollagen. The C-propeptide domains present at the C-terminal of the procollagen molecule promote the correct alignment of the chains. Once the triple helix formation is complete, procollagen is then transported from the ER to the Golgi apparatus for further modification and packaging into secretory vesicles.
When the extracellular space, the N and C terminal propeptides of the procollagen molecule are cleaved off by specific enzymes, transforming the molecule into tropocollagen. The tropocollagen molecules spontaneously align and aggregate to form collagen fibrils. These fibrils further organize to form collagen fibers.
The final step in collagen synthesis is the crosslinking of the collagen fibrils, a process catalyzed by the enzyme lysyl oxidase. This cross-linking contributes significantly to the tensile strength of collagen fibers. The intricate and tightly regulated process of collagen synthesis enables this crucial protein to provide structural support to various tissues in our body.
OI is a diverse genetic and clinical disease of the bones and connective tissues. It manifests itself in symptoms such as osteoporosis, brittle bones, unusually flexible joints, dentinogenesis imperfecta (DI), a blue tinge in the sclerae, and hearing loss that develops in adulthood. The variety of genetic defects and the ways they present themselves highlights the complex nature of OI. As such, the number of fractures and the extent of skeletal deformation can differ greatly between individuals or within affected families, potentially influenced by specific genes, genetic background, somatic mosaicism, or environmental factors.
In 1979, a new classification was introduced by Sillence et al., later expanded and refined by Levin and Shapiro. While this classification is beneficial, it does not fully encapsulate all cases of OI, especially since a third of cases are sporadic and two-thirds familial.
Pathophysiology of osteogenesis imperfecta
Osteogenesis Imperfecta (OI), often known as brittle bone disease, is a genetic disorder characterized by fragile bones that easily break. The pathophysiology of this disease is closely linked to abnormalities in the production of collagen, a protein that plays a critical role in maintaining the structural integrity of bones, skin, tendons, and other tissues.
In most cases of osteogenesis imperfecta, mutations occur in the genes (COL1A1 and COL1A2) that encode type I collagen. This form of collagen is the most abundant in bone tissue. These genetic abnormalities can lead to one of three outcomes: a reduction in the amount of type I collagen produced, the production of poor quality collagen, or a combination of both.
When the mutation results in fewer collagen being produced, the bones can become thin (low bone mass) and easily fracture. When the mutation leads to the production of poor quality collagen, the collagen structure is disorganized, resulting in bone fragility and deformities. The severity of the disease can vary greatly, ranging from mild cases to serious and potentially life-threatening conditions.
The most severe forms of Osteogenesis Imperfecta can present in utero with multiple fractures and bone deformities at birth. Other forms might not become apparent until later in life when the person begins to experience frequent fractures.
In addition to bone fragility, other clinical manifestations of osteogenesis imperfecta can include hearing loss, dental issues (dentinogenesis imperfecta), blue sclerae, short stature, and in severe cases, respiratory problems due to rib fractures and spine deformities.
Classification of Osteogenesis Imperfecta
Mild nondeforming OI (Sillence Type I)
This variant of OI constitutes about 60% of all OI patients. Bone fragility and osteoporosis are less severe compared to other types and generally do not result in skeletal deformity. Fractures are rarely present at birth (around 10%) and tend to occur once the child starts walking. The fracture rate decreases significantly decreases after puberty. Patients may show mild growth retardation and mild scoliosis.
Distinctive triangular-shaped facies are common among these patients and all possess blue sclerae. Dentinogenesis imperfecta (DI) is observed in 25% of cases, leading to a distinction between those with DI and those without DI.
A certain degree of joint laxity is common. About 70% of patients face hearing impairment, but only 10% require a hearing aid.
Mild OI follows an autosomal dominant pattern of inheritance. When it manifests itself as a new mutation, the phenotype is passed down as a dominant trait. There seems to be consistency in the inheritance of the disease, with marked variation in severity from the established family pattern being a rare occurrence.
Neonatal Lethal OI (Sillence Type II)
Neonatal lethal OI is the most intense form of this disease, leading to a substantial decrease in skeletal mass. Newborns with this form experience numerous fractures during gestation, resulting in short and malformed limbs, and may even suffer brain injury or dismemberment during birth.
Radiological observations typically reveal a misshapen large skull, wide and crushed extremities, a unique rib beading, and fractured vertebrae. However, there exists a range of radiological severity. These infants are often hypotonic, have feeding difficulties, and develop respiratory insufficiency shortly after birth, most succumbing to pulmonary failure within days or weeks.
Neonatal lethal OI affects approximately 10% of patients and approximately 20% of families. Instances of multiple affected siblings born to seemingly healthy parents have been reported, generally indicating recessive inheritance. The concept of new autosomal dominant mutations being a cause of fatal OI was introduced in 1970 and has been supported by recent biochemical evidence indicating that new mutations often cause this form of OI.
A comprehensive analysis of affected infants indicated that most cases occurred sporadically, with biochemical analysis of the parents strongly suggesting the abnormality in the infant as a new mutation. In families with more than one affected infant, pedigree and biochemical data most consistently point to one parent’s germinal mosaicism.
Severe nonlethal OI (Sillence Type III)
This variant of OI is identifiable at birth, with symptoms including fractures, long bone deformity, skull molding, and elongated pedicles of the vertebrae. The clinical course is marked by significant growth delay, frequent and deforming fractures of the long bones, and scoliosis coupled with chest wall deformity that can eventually result in respiratory insufficiency.
Those affected are typically wheelchair dependent. Joint laxity is more pronounced than in other nonfatal forms of OI. The sclerae usually appear white, juvenile, or senile, and hearing loss may manifest earlier due to increased cranial deformity.
Severe non-fatal OI affects around 20% of patients and 15% of families affected by OI. Most of these cases are sporadic, with no prior family history of bone disease. It is widely accepted that seemingly healthy parents can have multiple affected children, aligning with autosomal recessive inheritance. This OI type has been reported in nonidentical twins. However, most recurrent OI cases are now attributable to somatic mosaicism. Prenatal diagnosis of this OI type has been made in the second trimester, offering value for families with a previously affected individual.
Moderate and deforming OI (Sillence type IV)
This form of OI exhibits a broad array of characteristics, falling between severely deforming and mildly affected cases. As traditionally defined, a common feature among these individuals is the presence of blue sclerae in early life, which lighten during childhood and acquire normal color in adulthood. Those with white sclerae are a minority in intermediate OI cases. Interestingly, there have been observations of adult patients with moderately severe skeletal conditions typical of this OI phenotype, but still retaining blue sclerae. As such, it is suggested that classifying individual cases based on scleral color could be arbitrary and may not align with biochemical data.
Despite the challenge in determining the scleral color, there are other clinical features of this OI form. Typically, there is a moderate degree of growth retardation. Limb deformity often requires the use of braces or a cane for assistance walking. Severe scoliosis might occur. Joint laxity is common and about 20% require hearing aids due to hearing loss. Dentinogenesis imperfecta (DI) is frequently observed in this group, leading to a distinction between those with dental changes and those without.
Compared to type I OI, this phenotype is characterized by a higher rate of fractures at birth, but no significant difference in later years and a lower propensity for bruising. These patients also noted to develop more skeletal deformities than individuals with type I OI.
Genetic heterogeneity is also present in this group. Although the majority show autosomal dominant inheritance, the existence of sporadic cases implies the possibility of new mutations. Interestingly, an increase in paternal age does not seem to contribute to the occurrence of new mutations.
Treatment of Osteogenesis Imperfecta
The treatment options for Osteogenesis Imperfecta (OI) vary widely and depend on the severity and specific symptoms of the individual patient. The management of OI is mostly supportive and aims to increase the patient’s quality of life, reduce fracture rates, and optimize mobility and function.
Here are the primary treatment options:
- Bisphosphonates: These are the most commonly used drugs for OI. They work by slowing down the process of bone resorption, thereby helping to increase bone density and reduce the risk of fractures.
- Physical Therapy and Rehabilitation: Physical therapy plays a key role in the management of OI. Regular exercises help to strengthen muscles, improve balance and mobility, and maintain a healthy body weight. Rehabilitation services are also critical in managing the long-term physical disabilities associated with OI.
- Orthopedic Management: This is crucial for managing fractures and bone deformities in OI patients. It includes casting, rodding surgery (where metal rods are inserted into the long bones to provide support and prevent fractures), and physiotherapy.
- Mobility Aids: The use of wheelchairs, braces, or walking aids may be required to assist with mobility and independence.
- Dental Care: OI can sometimes affect the teeth (a condition known as dentinogenesis imperfecta), so regular dental check-ups and appropriate dental care are necessary.
- Hearing Management: As OI can be associated with hearing loss, particularly in adulthood, regular hearing checks are recommended. Hearing aids or other interventions may be required in some cases.
- Healthy Lifestyle: Maintaining a healthy lifestyle, including a nutritious diet and avoidance of smoking and excessive alcohol, is essential in the management of OI.
- Pain Management: Chronic bone pain and acute fracture pain are common in OI. Pain management strategies may include medications, physiotherapy, and cognitive-behavioral approaches.
- Psychosocial Support: Living with a chronic condition like OI can have significant psychological and emotional impacts. Thus, psychosocial support, including counseling and patient support groups, can be beneficial.
Reference
Sobczak-Kupiec A, Drabczyk A, Florkiewicz W, Głąb M, Kudłacik-Kramarczyk S, Słota D, Tomala A, Tyliszczak B. Review of the Applications of Biomedical Compositions Containing Hydroxyapatite and Collagen Modified by Bioactive Components. Materials. 2021; 14(9):2096.