Congenital adrenal hyperplasia (CAH) refers to a group of inherited enzymatic defects in cortisol biosynthesis. Impairment in cortisol production lifts the classic negative effect of cortisol on the hypothalamus and the pituitary gland.
Subsequently, this results in the secretion of corticotropin-releasing hormone (CRH) and ACTH, by the hypothalamus and anterior pituitary gland respectively, resulting in hyperplasia of the adrenal cortex. 21-hydroxylase deficiency (21OHD) also known as classic CAH accounts for almost 90% of CAH cases. Non classical adrenal hyperplasia is relatively less common and contributes to hyperandrogenemia.
Classification of Congenital adrenal hyperplasia
It is worth noting that 21OHD is grouped into classic and nonclassic forms, based on the presence or absence of cortisol insufficiency (adrenal insufficiency). Furthermore, the classic subtype of 21OHD is further dichotomized into “salt-wasting” and “simple virilizing” variants, depending on whether or not mineralocorticoid synthesis is sufficiently impaired.
The nonclassic 21OHD (NCCAH) is a much more frequent cause of clinical hyperandrogenemia, occurring in about 1 in 1,000 Caucasians. The prevalence is highly variable, depending on the ethnic group – Ashkenazi Jews (1:27), Hispanics (1:53), Yugoslavs (1:62), and Italians (1:300). The difference between the classic and non-classic forms is that girls who suffer from the latter do not develop either cortisol deficiency or overt clinical virilization (a sign of hyperandrogenemia).
Pathophysiology of Congenital adrenal hyperplasia
Steroidogenesis in the adrenal gland is dependent on the expression of enzymes in specific zones of the gland (review this simple mnemonic for remembering adrenal steroidogenesis here). This results in the synthesis of various adrenal steroids including aldosterone, cortisol, and dehydroepiandrosterone sulfate. The primary defect in CAH is an adrenal steroidogenic enzyme defect, which results in low levels of serum cortisol. The loss of negative feedback control activates the hypothalamic-pituitary-adrenal axis, which results in increased Adrenocorticotropic hormone (ACTH) production. ACTH stimulates the buildup and subsequent shunting of precursor steroids into either adrenal androgens or mineralocorticoid production[1]. Distinguishing between non classic and classic congenital adrenal hyperplasia? [2]
| Classic CAH | Non classic CAH (mild form of CAH) |
| Large gene deletion involving the CYP21A2 gene. | Usually, a point mutation (70% of patients) in the CYP21A2 gene. |
| Loss of 21-hydroxylase enzyme activity by 95-100% | Loss of 21-hydroxylase enzyme activity by 20-50% |
| Early onset (detected at birth) | Late onset (detected in the reproductive years) |
| Ambiguous genitalia in females | Normal genitals in females |
Other forms of CAH
The primary defect in CAH is an adrenal steroidogenic enzyme deficiency, which results in low levels of serum cortisol. The loss of negative feedback control activates the hypothalamic-pituitary-adrenal axis, which results in increased Adrenocorticotropic hormone (ACTH) production. ACTH stimulates the buildup and subsequent shunting of precursor steroids into either androgen or mineralocorticoid production[1]. The steroidogenic acute regulatory protein (StAR), at the level of the adrenal gland, mobilizes cholesterol from the outer to the inner mitochondrial membrane. The cytochrome P450 side-cleavage enzyme (P450scc) in the inner mitochondrial membrane converts cholesterol to pregnenolone; this is the rate-limiting step of adrenal steroidogenesis. The downstream effects of StAR are under trophic stimulation by both ACTH and luteinizing hormone (LH)[3]. Pregnenolone is then converted to progesterone by 3beta-hydroxysteroid dehydrogenase type 2 (HSD3B2), in the zona glomerulosa. Progesterone is then converted through a series of enzymatic steps involving 21 hydroxylase (CYP21A2) and aldosterone synthase into aldosterone.
In the zona fasciculata, pregnenolone is hydroxylated into 17-hydroxypregnenolone by CYP17A1 (17-alpha-hydroxylase enzyme/17,20 lyase). HSD3B2, CYP21A2, and CYP11B1 (11 beta-hydroxylase) are then involved in downstream reactions leading to the production of cortisol. In the zona reticularis, CYP17A1 and HSD3B2 are involved in the eventual formation of androgen precursors such as dehydroepiandrosterone (DHEA) and androstenedione[4].
Some of the types of CAH outlined below are rare disorders.
- Congenital lipoid hyperplasia (CLH). CLH is due to an autosomal recessive mutation in the gene encoding StAR. Clinical features include severe adrenal insufficiency (both mineralocorticoid and glucocorticoid deficiency) and external female genitalia in male infants. It is a severe form of CAH[5].
- 3beta-hydroxysteroid dehydrogenase type 2 deficiency. The clinical spectrum of 3BHSD varies from a presentation akin to CLH (both mineralocorticoid and glucocorticoid deficiency) to an even less severe subtype characterized by the absence of salt-wasting [6].
- 17 hydroxylase deficiency. A genetic mutation in CYP17A1 impairs steroidogenesis in both the adrenal glands and gonads[7]. Genetic females have normal-appearing female external genitalia at birth but subsequently present with delayed puberty. Male infants, on the other hand, are unable to synthesize testosterone, critical in developing male external genitalia. This results in the formation of female appearing external genitalia]8,9]. Also, males do not have internal Mullerian structures (uterus and fallopian tubes) since their anatomic testes continue to produce anti-Mullerian hormone. Despite patients being mildly glucocorticoid deficient, they seldom develop overt adrenal crises. This is because of the accumulation of precursors such as corticosterone and 11-deoxycorticosterone (DOC) not only activates the mineralocorticoid receptor to maintain blood pressure but also contributes to hypertension in affected patients[10].
- 11 beta-hydroxylase deficiency. A genetic mutation in CYP11B1 results in a defective 11beta hydroxylase, a critical enzyme in cortisol synthesis[11,12]. There is, therefore, an accumulation of proximal precursors such as DOC and 11-deoxycortisol. DOC has intrinsic mineralocorticoid activity and accounts for hypertension and hypokalemia observed in these patients[13]. There is an additional shunting of more proximal steroids into androgen production (DHEA, DHEA-S, and androstenedione). Genetic males have enlarged penile shafts at birth due to hyperandrogenemia. Genetic females have ambiguous genitalia due to exposure to elevated serum androgens during intrauterine life[13,14].
- 21 hydroxylase deficiency. This is a classical form of CAH – a genetic mutation in CYP21A2 results in a defective 21 hydroxylase enzyme. 21-hydroxylation of progesterone (mineralocorticoid synthesis pathway) and 17-hydroxyprogesterone (glucocorticoid synthesis pathway) is therefore impaired. There is a subsequent diversion of proximal steroidogenic precursors into androgen production. Newborns with more deleterious mutations in the CYP21A2 gene are hemodynamically unstable due to impaired glucocorticoid and mineralocorticoid synthesis. Genetic females present with ambiguous genitalia[15].
Clinical features of non classic CAH
Non classic CAH accounts for up to 2% of cases of hyperandrogenemia involving women in their reproductive years[16]. The clinical presentation of this condition is variable. A few clinical features include :
- Oligomenorrhea
- Infertility
- Hirsutism
- Acne
- Androgenic pattern of alopecia
- Acanthosis nigricans
- Acrochordons (skin tags)
- Testicular adrenal rest tumors (men)
Pathophysiology of the clinical features of non classic CAH
Hirsutism (excessive body hair) : Hirsutism, which is the growth of terminal male pattern hair, is evaluated with the modified Ferriman-Gallwey score, which happens to be a somewhat objective clinical assessment tool. There is wide variability in the prevalence of hirsutism, ranging from 1 to 33%[17].
The degree of hirsutism, however, does not correlate positively with circulating androgen levels due to differences in skin sensitivity to circulating androgens[1].
Pathophysiology
Most patients with NCCAH due to 21 hydroxylase deficiency typically have normal ACTH levels, in contrast to the high ACTH seen in the classic form. There is, however, excessive production of androgens in response to normal ACTH stimulation of the zona reticularis. This has been attributed to a missense mutation in the CYP21A2 gene, which results in a significant buildup of androgen precursor steroids, due to reduced activity of the 21-hydroxylase enzyme[18]. There is an increased production of metabolites, such as DHEA-S, androstenedione, and testosterone[19]. High circulating androgens account for the acne and hirsutism seen in NCCAH[18].
The elevated levels of adrenal steroids impair negative feedback control of progesterone on the hypothalamic-pituitary-ovarian axis. This causes hypersecretion of LH due to rapid GnRH pulse frequency, akin to what has been proposed for the polycystic ovarian syndrome (PCOS)[20]. LH stimulates the theca cells of the ovaries to increase ovarian androgen production[18,21].
Testicular adrenal rest tumors : Testicular adrenal rest tumors (TARTs) typically occur in males with classical congenital adrenal hyperplasia, although it has been reported in NCCAH as well[22]. TARTs in boys with congenital adrenal hyperplasia (CAH) are known to regress in size with optimal supplementation of hormonal insufficiencies. They are diagnosed based on clinical and radiographic findings, with biopsies being unnecessary in most cases[23].
Pathophysiology
Adrenal and gonadal cells are located adjacent to each other in the developing embryo. Inadvertent translocation of embryonic adrenal cells to the gonads occurs during gonadal descent. The lack of atrophy of these ectopic adrenal tissues in the setting of NCCAH or CAH is the cause of TARTs[23]. ACTH binds to ACTH receptors present on adrenal tissue in the testes, and this trophic stimulation of the ectopic adrenal tissue by ACTH results in their growth[24].
Clinical insulin resistance: Insulin resistance has a significant association with NCCAH[25,26], although some authors believe this association could be secondary to glucocorticoid use in this patient population[2,18]. Hyperinsulinemia-mediated skin manifestations have been described elsewhere.
Pathophysiology
The mechanisms underlying hyperinsulinemia in NCCAH are incompletely understood[26,27]. Normalization of serum androgen does not ameliorate insulin resistance, making hyperandrogenemia an unlikely cause of insulin resistance in patients with NCCAH[28].
Diagnosis of Congenital adrenal hyperplasia
Elevations of 17OHP, the main substrate of CYP21A2 (21 hydroxylase enzyme), are a hallmark of 21OHD, and 17 Hydroxyprogesterone (17OHP) has traditionally been used for both diagnosis and monitoring of the disease.
Measure baseline early morning 17OHP and an ACTH stimulated 17OHP level if baseline levels are fall between 200 to 1000 ng/dL. 17OHP levels below 200ng/dl (<6 nmol/L) is indicative of either a normal patient (unaffected by CAH) or a heterozygote mutation of the 21 hydroxylase enzyme. 17OHP levels above 1000ng/dl (>300 nmol/L) is diagnostic for classic CAH.
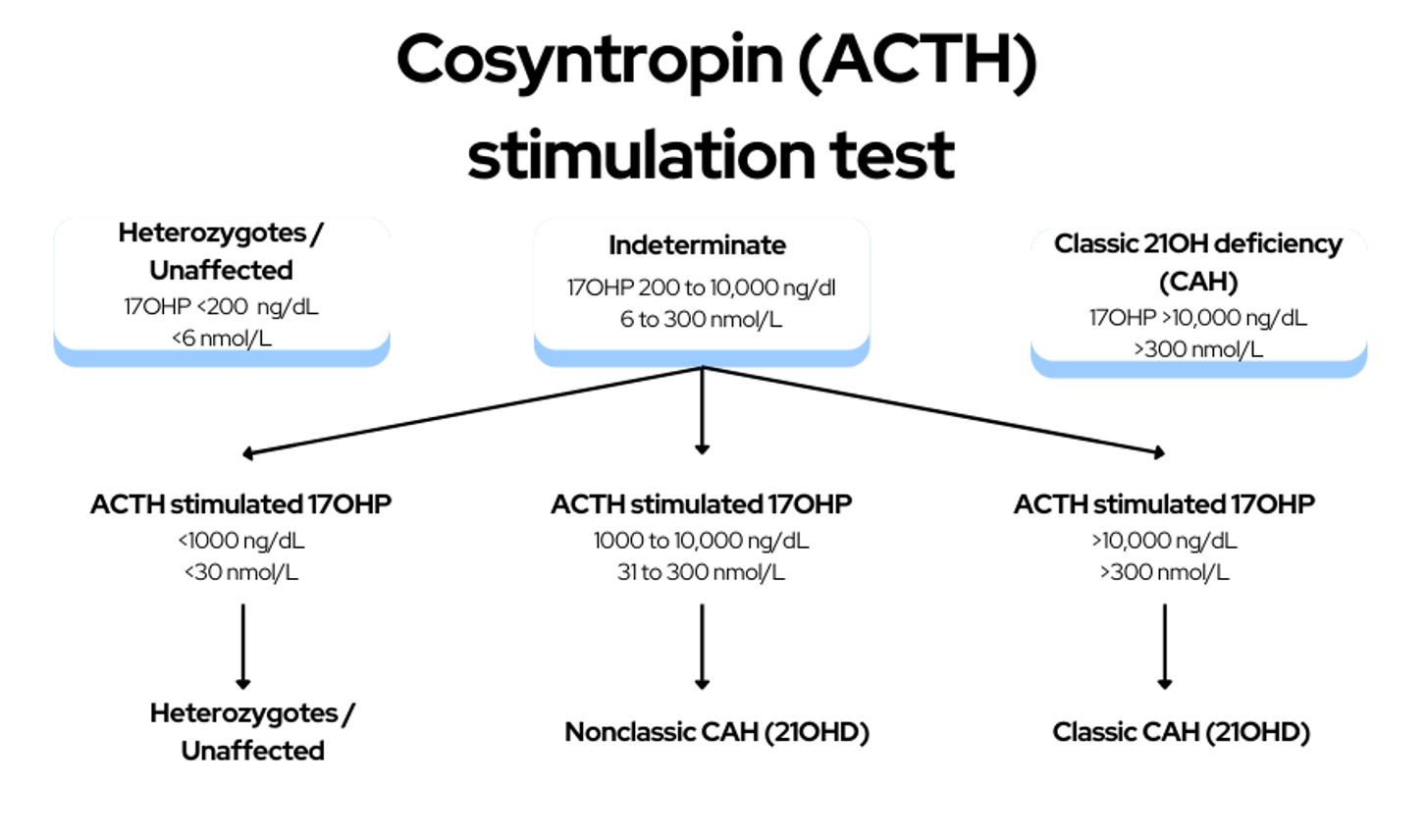
For patients with a baseline 17OHP level between 200 to 1000 ng/dL (6 to 300 nmol/L), a formal cortrosyn stimulation test is required.
Interpretation of the cosyntropin stimulation test
- 17 hydroxyprogesterone level of 1000 to 10,000 ng/dL (31 to 300 nmol/L) = Non classic congenital adrenal hyperplasia
- 17 hydroxyprogesterone level >10,000 ng/dL(>300 nmol/L) = Classic congenital adrenal hyperplasia
- 17 hydroxyprogesterone level <1000 ng/dL (<30 nmol/L) = Unaffected (or a heterozygous mutation of CYP21A2)
Medical Treatment of CAH
Treatment for classic 21-hydroxylase deficiency consists of glucocorticoid and mineralocorticoid replacement, and for both classic and non-classic disease, sufficient glucocorticoid is administered to correct the androgen excess. Patients with 21-hydroxylase deficiency are prone to developing adrenal cortical adenomas and myelolipomas, as well as adrenal rest tumors in the testis or elsewhere.
Glucocorticoids and mineralocorticoids are required in patients with 21OHD. Firstly, glucocorticoid therapy replaces deficient cortisol and also inhibits adrenal androgen overproduction. Glucocorticoids by exerting negative feedback on the hypothalamus and the pituitary, subsequently inhibit CRH and ACTH production respectively.
Management Pearls
Hydrocortisone is preferred in children and adolescents, until growth is completed, due to its short duration of action, which limits the potential to suppress growth. Hydrocortisone also serves as replacement therapy for adults, but long-acting synthetic glucocorticoids are often preferred, owing to less frequent dosing. The longer duration of action and higher potency of drugs like prednisolone and dexamethasone, however, might increase the risk of hypercortisolemia such as weight gain, dermal atrophy, and osteoporosis.
Similar to patients with primary adrenal insufficiency, stress doses of steroids should be given in patients with classic 21OHD during surgery, physical illness, labor and delivery. Exposure of the fetal brain to exogenous steroids can cause intellectual impairment. Dexamethasone crosses the placenta since it is not metabolized by placental 11 beta-hydroxysteroid dehydrogenase type 2(11BSD2). Prednisone and hydrocortisone, on the other hand, are metabolized by 11BHSD2 and, as such, preferred in pregnancy. For women in their reproductive years attempting to either conceive or are pregnant, a glucocorticoid that is inactivated by placental 11β-hydroxysteroid dehydrogenase type 2 (e.g., hydrocortisone, prednisone, and prednisolone) should be used, to avoid exposure the fetus to high doses of steroids.
Finally, asymptomatic patients with non classic congenital adrenal hyperplasia do not require glucocorticoid treatment, and stress doses of steroids are rarely needed.
Surgical Treatment of CAH
Bilateral adrenalectomy has been performed in selected patients with severe forms of 21OHD, in whom hyperandrogenism was difficult to control despite supraphysiologic glucocorticoid replacement. It is however worth noting that primary adrenal insufficiency, which subsequently develops is also very difficult to manage. It certainly involves lifelong adherence to both glucocorticoid and mineralocorticoid replacement, in order to prevent a potentially life threatening adrenal crisis.
References
1. Witchel SF. Congenital Adrenal Hyperplasia. J Pediatr Adolesc Gynecol. 2017;30(5):520-534. doi:10.1016/j.jpag.2017.04.001
2. Trapp CM, Oberfield SE. Recommendations for treatment of nonclassic congenital adrenal hyperplasia (NCCAH): An update. Steroids. 2012;77(4):342-346. doi:10.1016/j.steroids.2011.12.009
3. King SR, Stocco DM. Steroidogenic Acute Regulatory Protein Expression in the Central Nervous System. Front Endocrinol. 2011;2. doi:10.3389/fendo.2011.00072
4. Al Alawi AM, Nordenström A, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine. 2019;63(3):407-421. doi:10.1007/s12020-018-01835-3
5. Kim CJ. Congenital lipoid adrenal hyperplasia. Ann Pediatr Endocrinol Metab. 2014;19(4):179-183. doi:10.6065/apem.2014.19.4.179
6. Sahakitrungruang T. Clinical and molecular review of atypical congenital adrenal hyperplasia. Ann Pediatr Endocrinol Metab. 2015;20(1):1-7. doi:10.6065/apem.2015.20.1.1
7. Fontenele R, Costa-Santos M, Kater CE. 17α-hydroxylase deficiency is an underdiagnosed disease: high frequency of misdiagnosis in a large cohort of Brazilian patients. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol. 2018;24(2):170-178. doi:10.4158/EP171987.OR
8. Kim SM, Rhee JH. A case of 17 alpha-hydroxylase deficiency. Clin Exp Reprod Med. 2015;42(2):72-76. doi:10.5653/cerm.2015.42.2.72
9. Kardelen AD, Toksoy G, Baş F, et al. A Rare Cause of Congenital Adrenal Hyperplasia: Clinical and Genetic Findings and Follow-up Characteristics of Six Patients with 17-Hydroxylase Deficiency Including Two Novel Mutations. J Clin Res Pediatr Endocrinol. 2018;10(3):206-215. doi:10.4274/jcrpe.0032
10. Auchus RJ. Steroid 17-Hydroxylase and 17,20-Lyase Deficiencies, Genetic and Pharmacologic. J Steroid Biochem Mol Biol. 2017;165(Pt A):71-78. doi:10.1016/j.jsbmb.2016.02.002
11. White PC. Congenital adrenal hyperplasia owing to 11β-hydroxylase deficiency. Adv Exp Med Biol. 2011;707:7-8. doi:10.1007/978-1-4419-8002-1_2
12. Ben Charfeddine I, Riepe FG, Kahloul N, et al. Two novel CYP11B1 mutations in congenital adrenal hyperplasia due to steroid 11β hydroxylase deficiency in a Tunisian family. Gen Comp Endocrinol. 2012;175(3):514-518. doi:10.1016/j.ygcen.2011.12.017
13. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine. 2017;55(1):19-36. doi:10.1007/s12020-016-1189-x
14. Khattab A, Haider S, Kumar A, et al. Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2017;114(10):E1933-E1940. doi:10.1073/pnas.1621082114
15. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21(3):245-291. doi:10.1210/edrv.21.3.0398
16. Ambroziak U, Kępczyńska-Nyk A, Kuryłowicz A, et al. The diagnosis of nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency, based on serum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf). 2016;84(1):23-29. doi:10.1111/cen.12935
17. Witchel SF, Azziz R. Nonclassic Congenital Adrenal Hyperplasia. Int J Pediatr Endocrinol. 2010;2010. doi:10.1155/2010/625105
18. Carmina E, Dewailly D, Escobar-Morreale HF, et al. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update. 2017;23(5):580-599. doi:10.1093/humupd/dmx014
19. Speiser PW, Arlt W, Auchus RJ, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(11):4043-4088. doi:10.1210/jc.2018-01865
20. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. 2013;78(8):747-750. doi:10.1016/j.steroids.2013.04.010
21. Blank SK, McCartney CR, Helm KD, Marshall JC. Neuroendocrine Effects of Androgens in Adult Polycystic Ovary Syndrome and Female Puberty. Semin Reprod Med. 2007;25(05):352-359. doi:10.1055/s-2007-984741
22. Kurtoğlu S, Hatipoğlu N. Non-Classical Congenital Adrenal Hyperplasia in Childhood. J Clin Res Pediatr Endocrinol. 2017;9(1):1-7. doi:10.4274/jcrpe.3378
23. Kocova M, Janevska V, Anastasovska V. Testicular adrenal rest tumors in boys with 21-hydroxylase deficiency, timely diagnosis and follow-up. Endocr Connect. 2018;7(4):544-552. doi:10.1530/EC-18-0097
24. Kim MS, Goodarzian F, Keenan MF, et al. Testicular Adrenal Rest Tumors in Boys and Young Adults with Congenital Adrenal Hyperplasia. J Urol. 2017;197(3 Pt 2):931-936. doi:10.1016/j.juro.2016.09.072
25. Powell D, Inoue T, Bahtiyar G, Fenteany G, Sacerdote A. Treatment of Nonclassic 11-Hydroxylase Deficiency with Ashwagandha Root. Case Reports in Endocrinology. doi:10.1155/2017/1869560
26. Saygili F, Oge A, Yilmaz C. Hyperinsulinemia and insulin insensitivity in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency: the relationship between serum leptin levels and chronic hyperinsulinemia. Horm Res. 2005;63(6):270-274. doi:10.1159/000086363
27. Pall M, Azziz R, Beires J, Pignatelli D. The phenotype of hirsute women: a comparison of polycystic ovary syndrome and 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil Steril. 2010;94(2):684-689. doi:10.1016/j.fertnstert.2009.06.025
28. Singer F, Bhargava G, Poretsky L. Persistent insulin resistance after normalization of androgen levels in a woman with congenital adrenal hyperplasia. A case report. J Reprod Med. 1989;34(11):921-922.