A detailed review of a classification system for the various pathophysiologic defects in hyperlipidemia. Fredrickson’s classification is, however, an oversimplification of the multiple causes of hyperlipidemia.
Type I hyperlipoproteinemia (hyperlipidemia)
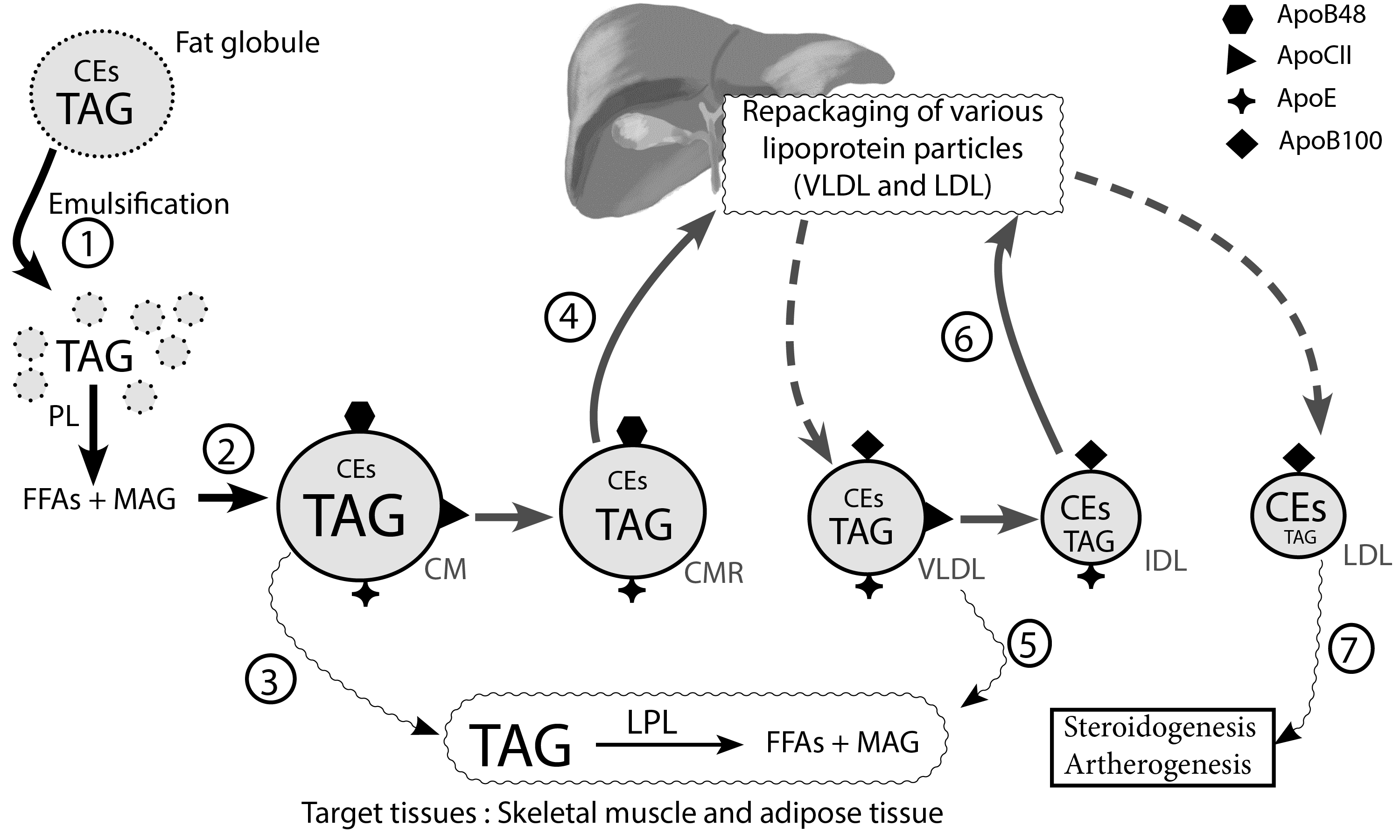
Deficiencies of either lipoprotein lipase (LPL)[1] or apolipoprotein CII (Apo-CII) have been reported[2]. These defects result in an elevated level of triglyceride-rich chylomicrons.
Clinical features include recurrent pancreatitis, lipemia retinalis, tubero-eruptive xanthomas, and hepatosplenomegaly[3].
We will refer readers to step 3 of the lipoprotein synthesis pathway. Note the importance of Apo CII and lipoprotein lipase in the hydrolysis of triglyceride-laden chylomicrons.
 Cholesterol Synthesis Pathway
Cholesterol Synthesis Pathway
Type IIa hyperlipoproteinemia (Familial hypercholesterolemia)
Mutations in the LDL receptor (LDL-R) and apolipoprotein B (ApoB) genes have been reported[4], with mutations of the LDL-R accounting for 85-90% of cases[5]. Activating mutations in proprotein convertase subtilisin kexin type 9 (PCSK9), an enzyme complex involved in the recycling of the LDL-R, limits the lifespan of LDL-R and is responsible for a small subset of patients with type IIa hyperlipoproteinemia[5].
There is an elevated level of total cholesterol and LDL. HDL and TAGs are usually normal [5]. Clinical features include corneal arcus, xanthelasma, or tuberous xanthomas[5].
Type IIb hyperlipoproteinemia (familial combined hyperlipidemia)
It is the most common genetic cause of dyslipidemia in the general population. The primary defect in type IIb hyperlipidemia is yet to be elucidated[6], it, however, leads to excessive production of VLDL (triglyceride-laden lipoprotein)[7] and LDL[8, 9].
This clinical phenotype fits the typical “atherogenic lipid triad,” which consists of an elevated serum concentration of small dense LDL with high TAGs, high apolipoprotein B, and reduced HDL[7].
Physical findings suggestive of dyslipidemia are uncommon in familial combined hyperlipidemia[10]. It is worthy to note that the most frequent cause of dyslipidemia in the general population seldom has any of the physical manifestations we discussed earlier in this chapter.
Type III hyperlipoproteinemia (dysbetalipoproteinemia)
Elevated VLDL remnants (IDL) and chylomicrons account for the high levels of total cholesterol and triglycerides in these patients[11]. There is a defect in the processing of chylomicrons and VLDL by the liver, which results in an increased circulating half-life of VLDL and chylomicrons.
CETP, therefore, has an opportunity to transfer more cholesterol esters from HDL to CM and VLDL due to the high levels of the latter two lipoproteins in circulation. Elevated concentrations of these abnormal lipoproteins result in high blood cholesterol: triglyceride ratio[11].
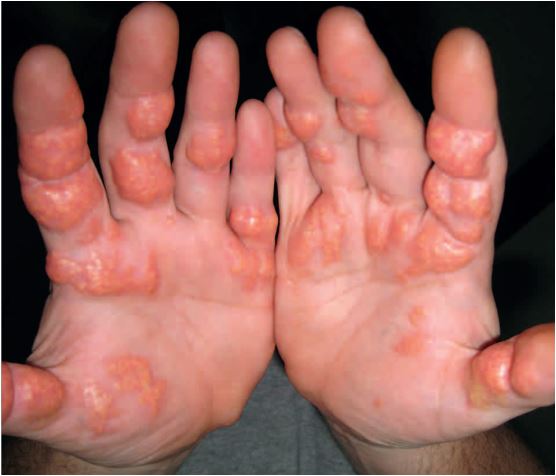
Figure 1. Palmar xanthomas. Palmar xanthomas appear as yellowish-orange macules with a predilection for the palmar creases[12]. They are pathognomonic for Type III hyperlipoproteinemia (familial dysbetalipoproteinemia) [13] and may, on rare occasions, occur in patients with familial hypercholesterolemia[14]. PX should be distinguished from planar xanthomas, which are whitish plaques and tend to extend beyond the confines of the palmar creases[12]. Palmar xanthomas involving the palmar creases are classically referred to as xanthoma striatum palmare[15]. Image source : Pathognomonic Palmar Crease Xanthomas of Apolipoprotein E2 Homozygosity-Familial Dysbetalipoproteinemia
Type IV hyperlipoproteinemia (simple hypertriglyceridemia)
- An isolated elevation of VLDL characterizes simple hypertriglyceridemia. Unlike the monogenic inheritance pattern of familial hypercholesterolemia, Type IV hyperlipoproteinemia is instead mostly polygenic in its etiology[10]. There is an elevated level of mainly VLDL-derived plasma triglycerides [10] and concomitant lowering of HDL cholesterol[16].
- Clinical features include eruptive xanthomas, lipemia retinalis, and hepatosplenomegaly [10]. Type IV hyperlipoproteinemia seldom causes acute pancreatitis except during specific metabolic stresses, when the Type IV phenotype can change to a Type V phenotype with both increased VLDL and chylomicrons[10]
- This form of hyperlipoproteinemia is sometimes called “diabetic dyslipidemia” as it is frequently encountered in people with type 2 diabetes mellitus (T2DM), and is considered part of the metabolic (insulin resistance) syndrome[16]. It is also associated with cardiovascular disease[17] and non-alcoholic fatty liver disease[18].
Type V hyperlipoproteinemia
- The etiology is multifactorial, including genetic mutations that result in altered triglyceride metabolism. Acquired factors such as alcohol, uncontrolled diabetes, steroids, estrogens, and medications play contributory roles in type V hyperlipoproteinemia. It is characterized by VLDL and chylomicron elevation, which results in an elevation of both triglycerides and total cholesterol[19]. The clinical features are similar to those of type 1 hyperlipoproteinemia[10]. Unlike familial hyperchylomicronemia, patients with Type V hyperlipoproteinemia are at risk for cardiovascular disease[20–22].
- There is evidence that although large chylomicrons are unable to penetrate the vasculature, VLDL and smaller remnants of chylomicrons can disrupt the integrity of the vascular endothelium. This sets off an inflammatory cascade that culminates in significant artherogenesis[23]. A useful mnemonic for remembering the features of this type of dyslipidemia is 1+4=5 because it has the combined features of Types I and IV hyperlipoproteinemia.
Tangier disease
- Tangier disease is named after the island in Chesapeake Bay, from which the first reported cases hailed [24]. It is caused by a genetic mutation in the ATP-binding cassette transporter A1 (ABCA1) gene, a gene that encodes the critical regulatory membrane transporter ABCA1. ABCA1 mediates the transfer of free cholesterol from extrahepatic tissues to apoA-1, to form the nascent HDL particle[25].
- It is a severe form of “hypolipoproteinemia,” which affects HDL metabolism. Patients have very low serum HDL, low total cholesterol with normal or even high plasma triglycerides[25]. The clinical features include yellowish-orange discoloration of the tonsils, tonsillar enlargement, hepatosplenomegaly, and peripheral neuropathy[24].
- The defect in ABCA1-mediated mobilization of peripheral cholesterol leads to the accumulation of lipids in various tissues, including the spleen, nerves, skin, and lymphoid tissue. Indeed, the accumulation of lipids in neurons leads to their devitalization and eventual demyelination.
Familial LCAT Deficiency
Lecithin cholesterol acyltransferase is a regulatory enzyme required for the esterification of peripherally acquired free cholesterol into cholesterol esters in the nascent HDL particle[26]. It is a rare condition with about 100 case reports to date[27].
Familial LCAT deficiency is due to mutations in the CAT gene, which results in reduced esterification of free cholesterol into cholesterol esters.
Patients with LCAT deficiency, therefore, have a high free cholesterol to esterified cholesterol ratio[28, 29]. Subjects with partial LCAT deficiency classically present with extensive corneal opacification that is often referred to as “fish-eye disease” (FED). Complete LCAT deficiency presents with a triad of corneal opacification (FED), anemia, and renal dysfunction[30].
Classification of the various forms of hyperlipidemia
A summary of the various clinical phenotypes of hyperlipoproteinemias is based on Dr. Donald S. Fredrickson’s classification, which was first published in the journal Circulation in 1965[31]. The classification system is an oversimplification of lipoprotein metabolism but is a good starting point in terms of appreciating the various forms of hyperlipoproteinemias.
Table 1 Fredrickson classification of hyperlipoproteinemias
| Type | Primary hyperlipidemia classification | Elevated lipoprotein |
| I | Familial hyperchylomicronemia syndrome | Chylomicrons[3] |
| IIa | Familial hypercholesterolemia | LDL[5] |
| IIb | Familial combined hypercholesterolemia | LDL, VLDL[8] |
| III | Familial dysbetalipoproteinemia | IDL[9, 11] |
| IV | Simple hypertriglyceridemia | VLDL[10] |
| V | Familial hypertriglyceridemia | Chylomicrons, VLDL[19] |
Adapted from references [3, 5, 8–11, 19]
References
- Pingitore P, Lepore SM, Pirazzi C, et al (2016) Identification and characterization of two novel mutations in the LPL gene causing type I hyperlipoproteinemia. J Clin Lipidol 10:816–823
- Wolska A, Dunbar RL, Freeman LA, Ueda M, Amar MJ, Sviridov DO, Remaley AT (2017) Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 267:49–60
- Patni N, Quittner C, Garg A (2018) Orlistat Therapy for Children With Type 1 Hyperlipoproteinemia: A Randomized Clinical Trial. J Clin Endocrinol Metab 103:2403–2407
- Saint-Jore B, Varret M, Dachet C, et al (2000) Autosomal dominant type IIa hypercholesterolemia: evaluation of the respective contributions of LDLR and APOB gene defects as well as a third major group of defects. Eur J Hum Genet 8:621–630
- Pejic RN (2014) Familial Hypercholesterolemia. Ochsner J 14:669–672
- Ellis KL, Pang J, Chan DC, Hooper AJ, Bell DA, Burnett JR, Watts GF (2016) Familial combined hyperlipidemia and hyperlipoprotein(a) as phenotypic mimics of familial hypercholesterolemia: Frequencies, associations and predictions. J Clin Lipidol 10:1329-1337.e3
- Arai H, Ishibashi S, Bujo H, et al (2012) Management of type IIb dyslipidemia. J Atheroscler Thromb 19:105–114
- Joerger M, Riesen WF, Thürlimann B (2011) Bevacizumab-associated hyperlipoproteinemia type IIb in a patient with advanced invasive-ductal breast cancer. J Oncol Pharm Pract 17:292–294
- Hegele RA, Ban MR, Hsueh N, Kennedy BA, Cao H, Zou GY, Anand S, Yusuf S, Huff MW, Wang J (2009) A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia. Hum Mol Genet 18:4189–4194
- Brahm A, Hegele RA (2013) Hypertriglyceridemia. Nutrients 5:981–1001
- Sniderman AD, de Graaf J, Thanassoulis G, Tremblay AJ, Martin SS, Couture P (2018) The spectrum of type III hyperlipoproteinemia. J Clin Lipidol 12:1383–1389
- Rothschild M, Duhon G, Riaz R, Jetty V, Goldenberg N, Glueck CJ, Wang P (2016) Pathognomonic Palmar Crease Xanthomas of Apolipoprotein E2 Homozygosity-Familial Dysbetalipoproteinemia. JAMA Dermatol 152:1275–1276
- Sharma D, Thirkannad S (2010) Palmar xanthoma-an indicator of a more sinister problem. Hand (N Y) 5:210–212
- Daroach M, Mahajan R Palmar crease xanthomas in familial hypercholesterolemia. Int J Dermatol 58:491–492
- Shenoy C, Shenoy MM, Rao GK (2015) Dyslipidemia in Dermatological Disorders. N Am J Med Sci 7:421–428
- Schofield JD, Liu Y, Rao-Balakrishna P, Malik RA, Soran H (2016) Diabetes Dyslipidemia. Diabetes Ther 7:203–219
- Rodriguez V, Newman JD, Schwartzbard AZ (2018) Towards more specific treatment for diabetic dyslipidemia. Curr Opin Lipidol 29:307–312
- Amor AJ, Perea V (2019) Dyslipidemia in nonalcoholic fatty liver disease. Current Opinion in Endocrinology, Diabetes and Obesity 26:103
- Gotoda T, Shirai K, Ohta T, et al (2012) Diagnosis and management of type I and type V hyperlipoproteinemia. J Atheroscler Thromb 19:1–12
- Sniderman AD, Couture P, Martin SS, DeGraaf J, Lawler PR, Cromwell WC, Wilkins JT, Thanassoulis G (2018) Hypertriglyceridemia and cardiovascular risk: a cautionary note about metabolic confounding. J Lipid Res 59:1266–1275
- Tenenbaum A, Klempfner R, Fisman EZ (2014) Hypertriglyceridemia: a too long unfairly neglected major cardiovascular risk factor. Cardiovascular Diabetology 13:159
- Paquette M, Bernard S, Hegele RA, Baass A (2019) Chylomicronemia: Differences between familial chylomicronemia syndrome and multifactorial chylomicronemia. Atherosclerosis 283:137–142
- Han SH, Nicholls SJ, Sakuma I, Zhao D, Koh KK (2016) Hypertriglyceridemia and Cardiovascular Diseases: Revisited. Korean Circ J 46:135–144
- Rader DJ, deGoma EM (2012) Approach to the Patient with Extremely Low HDL-Cholesterol. J Clin Endocrinol Metab 97:3399–3407
- Puntoni M, Sbrana F, Bigazzi F, Sampietro T (2012) Tangier disease: epidemiology, pathophysiology, and management. Am J Cardiovasc Drugs 12:303–311
- Dullaart RPF, Perton F, Sluiter WJ, de Vries R, van Tol A (2008) Plasma Lecithin: Cholesterol Acyltransferase Activity Is Elevated in Metabolic Syndrome and Is an Independent Marker of Increased Carotid Artery Intima Media Thickness. J Clin Endocrinol Metab 93:4860–4866
- Norum KR (2017) The function of lecithin:cholesterol acyltransferase (LCAT). Scand J Clin Lab Invest 77:235–236
- McIntyre N (1988) Familial LCAT deficiency and fish-eye disease. J Inherit Metab Dis 11 Suppl 1:45–56
- Morales E, Alonso M, Sarmiento B, Morales M (2018) LCAT deficiency as a cause of proteinuria and corneal opacification. BMJ Case Rep. https://doi.org/10.1136/bcr-2017-224129
- Kanai M, Koh S, Masuda D, Koseki M, Nishida K (2018) Clinical features and visual function in a patient with Fish-eye disease: Quantitative measurements and optical coherence tomography. Am J Ophthalmol Case Rep 10:137–141
- Fredrickson DS, Lees RS (1965) A system for phenotyping hyperlipoproteinemia. Circulation 31:321–327