Acromegaly is a chronic disorder that results when the pituitary gland produces excess growth hormone (GH). This excess GH leads to abnormal growth of the hands and feet, bones of the face, and internal organs. In children, excess GH leads to gigantism. Most cases of acromegaly are caused by a pituitary adenoma, a benign tumor of the pituitary gland. Less commonly, acromegaly is caused by pituitary hyperplasia, an overgrowth of normal pituitary cells, or certain rare disorders such as MEN type 1 or McCune-Albright syndrome.
In some cases, the cause of acromegaly is unknown. While acromegaly can occur in men and women, it is more common in women. Treatment for acromegaly generally involves surgery to remove the pituitary adenoma, radiation therapy, or medication. If left untreated, acromegaly can lead to serious health problems like diabetes, hypertension, sleep apnea, and heart disease. However, with early diagnosis and proper treatment, most people with acromegaly can live normal, healthy lives.
Introduction
The clinical syndrome of acromegaly (in adults) occurs due to a pituitary tumor’s chronic growth hormone production. This same condition is known as pituitary gigantism in a child or adolescent prior to the fusion of the epiphyseal growth plates. Pituitary gigantism thus results in progressive linear growth, resulting in an affected individual attaining an incredible height. Indeed cases of individuals with pituitary gigantism measuring almost 9 feet in height have been reported.
In contrast, acromegaly occurs after the fusion of the growth plates (adults), and thus, there is no progressive linear growth. However, acromegalics experience peculiar acral (refers to the extremities) changes diagnostic of growth hormone excess.
Acromegaly has a prevalence of approximately 3 per million persons in the population. Indeed it is second only to prolactinomas as a common cause of pituitary hyperfunction.
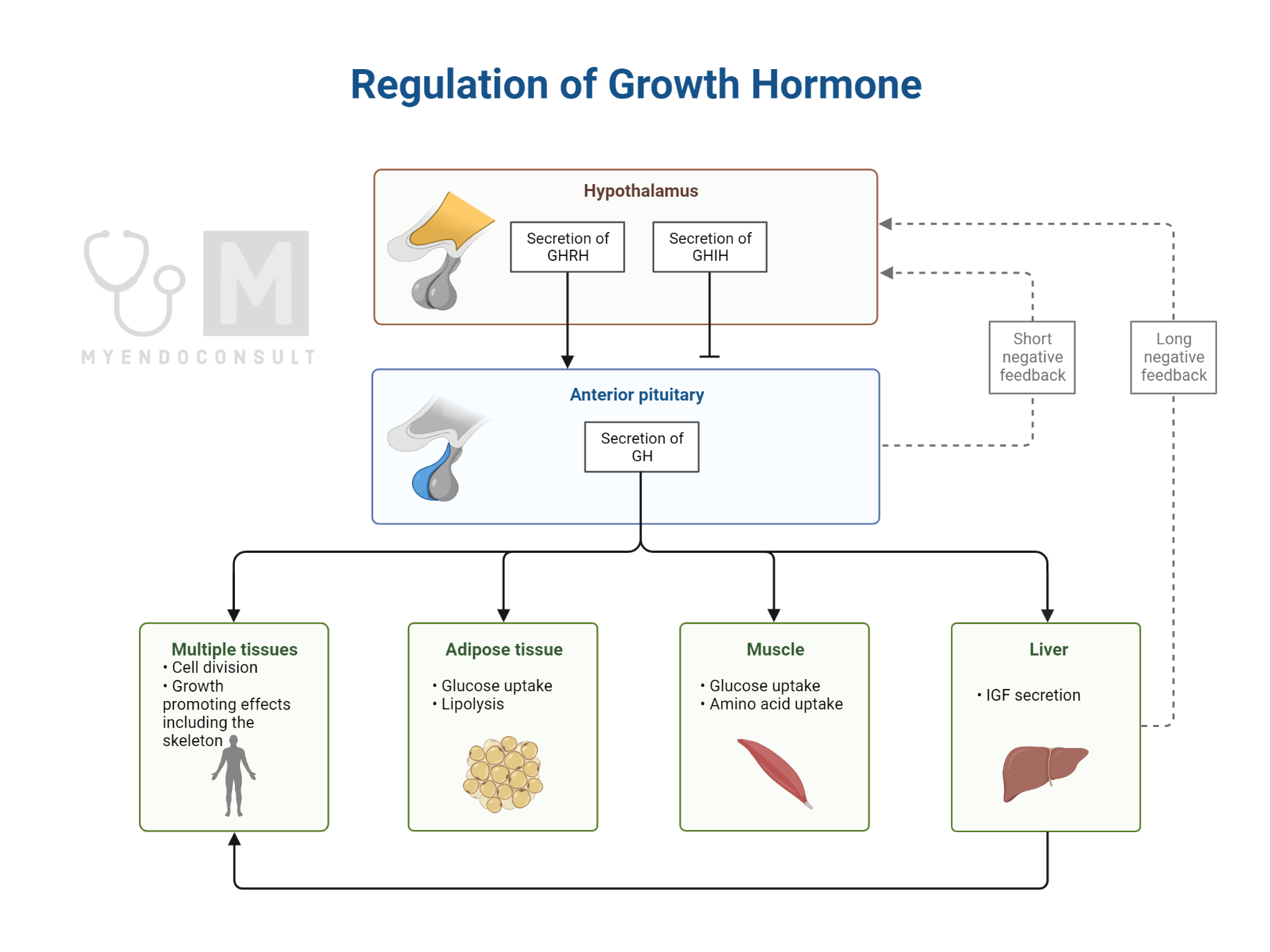
Pathophysiology of Acromegaly
Acromegaly is an endocrine disorder characterized by excessive secretion of growth hormone (GH) by somatotrophs. GH is a peptide hormone secreted by the pituitary gland, and it plays an important role in regulating body growth and development. The release of excess GH results in the overproduction of insulin-like growth factor 1 (IGF-1), which leads to changes in bone and soft-tissue growth.
Etiology of acromegaly
The causes of growth hormone excess can be grouped into conditions associated with primary growth hormone excess (involving the pituitary gland itself), extra pituitary growth hormone excess, or growth hormone-releasing hormone excess (GHRH).
Primary growth hormone excess
- Pituitary adenoma
- Pituitary carcinoma
- Extrapituitary location of somatotrophs (embryonic anomaly)
- Familial syndromes (Multiple endocrine neoplasia type 1, Carney syndrome, Familial acromegaly, and McCune-Albright syndrome)
Extrapituitary growth hormone excess
- Pancreatic neuroendocrine tumor (islet cell origin)
- Lymphomas
- Iatrogenic or even surreptitious GH abuse by athletes
Growth hormone-releasing hormone excess
- Hypothalamic tumor involving GHRH producing cells
- Release of GHRH by various neuroendocrine cells (bronchial carcinoids, pancreatic islet cell tumors, small cell lung cancer, pheochromocytomas, and medullary thyroid carcinoma)
Clinical Features
History
Patients may present with visual field complaints, headaches, proximal muscle weakness, oily skin, sleep apnea, narcolepsy, and enlargement of acral regions (hands and feet). Reported change in shoe or ring size suggests acral enlargement.
Clinical examination
Patients have coarse facial features, frontal bossing, spadelike hands, wide feet, and prognathism.
Additional features of the physical examination
- Hypertension
- Visceromegaly (enlarged tongue, thyroid gland, salivary glands, liver, spleen, prostate)
- Oily skin
- Skin tags
- Jaw malocclusion
- Cranial nerve palsies (mainly involving components nerves in the cavernous sinus)
- Wide interdental space
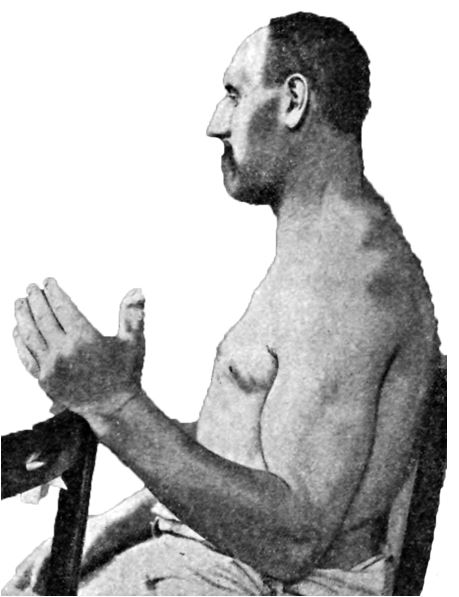 Clinical features of acromegaly
Clinical features of acromegaly
Investigations
Laboratory tests
Growth hormone is released intermittently, while insulin-like growth factor 1 (IGF-1) tends not to fluctuate (steady plasma concentration) in normal physiology. It is also worthy to note that IGF-1 does decline with age. Elevated serum IGF-1 levels are rarely false positives, although pregnancy can result in levels 2-3x the upper limit of normal.
Diagnostic criteria for acromegaly
- Failure of growth hormone to suppress to less than 0.4ng/dL (for ultrasensitive assays) or <1ng/dL (for older assays) within 2 hours of an oral glucose load (75g of anhydrous glucose)
- An elevated serum IGF-1 level (adjusted for age and gender-matched nomogram)
Supportive investigations
- Hypertriglyceridemia
- Impaired glucose tolerance
- Hypercalciuria
- Increased levels of 25 hydroxyvitamin D3
- Low renin, high aldosterone levels
- Low sex hormone-binding globulin
- Low thyroxine-binding globulin
Further evaluation after confirmation of diagnosis
- Serum calcium, phosphorus
- Blood glucose
- Fasting lipid profile
- Free T4 , TSH, 0800 h cortisol, testosterone/estradiol, and prolactin
- EKG
- Colonoscopy at baseline in ALL patients with acromegaly
- DXA (Bone densitometry) in hygogonadal patients
Imaging
MRI of the pituitary gland is routinely used in the evaluation of patients with acromegaly. This modality provides excellent anatomic detail of the sella turcica, allowing for precise characterization of pituitary adenomas.
MRI can also help to assess for other important findings in acromegaly including increased intrasellar pressure, which may be seen as dilation of the third ventricle and/or specks of gadolinium within the sella on T1-weighted images. Furthermore, MRI can sometimes reveal extrasellar extension of the adenoma, which may be important to consider when planning surgery.
A pituitary MRI diagnosis most causes of acromegaly, with 75-80% being macroadenomas and 20-25% being microadenomas.
Surgical Treatment of Acromegaly
Transsphenoidal surgery is successful in achieving remission in 90% of patients with microadenomas and 50% of patients with macroadenomas. Tumor size is the main predictor of remission after transsphenoidal surgery. Patients with small tumors are more likely to achieve remission than those with large tumors.
Predicting remission of acromegaly after surgery
The growth hormone–glucose tolerance test (GH-GTT) can be done as early as 1 week postoperatively and nadir levels below 1 ng/mL are predictive of long-term remission in up to 98% of patients. Also, a postoperative day 1 fasting serum GH level <2 ng/mL is predictive of sustained clinical and biochemical remission at 5 years.
| Post operative status | Features |
| Surgical Cure | Normalization of age-adjusted IGF-1 level Random GH <1ng/mL Suppressed GH after a 75g GTT Preservation of other anterior pituitary function Complete removal of the tumor |
| Active Disease | Elevated age-adjusted IGF-1 level Random GH >1ng/mL Elevated GH after a 75g GTT Persistence of the tumor |
Medical Treatment of Acromegaly
The indications for medical treatment of acromegaly include patients who are deemed high risk for transsphenoidal surgery, and invasive macroadenoma with parasellar extension but no mass effect. Also, patients with McCune-Albright syndrome either due to the absence of an anatomic lesion (remember, this occurs as a result of constitutive activation of the Gs alpha subunit of the GPCR) or presence of significant cranial fibrous dysplasia. Due to the high remission rates after surgery for those with microadenomas, medical therapy should not be selected as the primary treatment in this patient subset.
GH levels are normally regulated by somatostatin, which binds to and inhibits GH release. However, in patients with acromegaly, somatostatin levels are frequently insufficient to control GH production. As a result, somatostatin analogs are often prescribed in an attempt to control GH levels.
Somatostatin receptor ligands are the most common medical treatment for acromegaly. These drugs bind to and activate somatostatin receptors, which leads to a decrease in growth hormone secretion. The most commonly used somatostatin receptor ligands are octreotide and lanreotide. Pasireotide is a newer somatostatin receptor ligand that is also effective in some cases.
Somatostatin analogs (SSAs) are a class of drugs that bind to and activate somatostatin receptors (SSTRs). Somatostatin is a naturally occurring hormone that inhibits the secretion of growth hormone (GH) from the somatotroph cells in the pituitary gland. The use of SSAs for the treatment of acromegaly was first reported in the early 1980s. Since then, several SSAs have been developed and are now used clinically. The currently available SSAs include octreotide (octreotide short release), somatuline (lanreotide), and sandostatin (octreotide long-acting release). All three of these agents are approved by the United States Food and Drug Administration (FDA) for the treatment of acromegaly.
The mechanism of action of SSAs in the treatment of acromegaly is thought to be mediated through their ability to bind to and activate SSTRs on somatotroph cells. This leads to the inhibition of GH secretion from these cells and, consequently, a reduction in serum IGF-1 levels.
What is somatostatin resistance?
Unfortunately, some patients do not respond adequately to somatostatin analog therapy, either because their somatostatin receptors are resistant to the ligands or because of other factors. This resistance is defined as failure to reduce GH and IGF1 levels by 50% or tumor shrinkage by 20% after one year of treatment. While the exact prevalence of somatostatin analog resistance is unknown, it is thought to be fairly common, affecting up to 40% of patients with acromegaly.
There is currently no cure for somatostatin analog resistance, but various strategies are being investigated in an attempt to improve outcomes in these patients. There are several potential mechanisms of somatostatin analog resistance, including decreased somatostatin receptor expression and somatostatin receptor mutations.
Pegvisomant is a relatively new medication used in the treatment of acromegaly, pegvisomant’s mechanism of action is unique in that it targets the growth hormone receptor (GHR) directly. pegvisomant structurally mimics growth hormone (GH), allowing it to bind and occupy the GH receptor pocket. This prevents GH from binding and activating the GHR, thus halting the downstream signaling cascade that leads to cell proliferation. Additionally, pegvisomant does not activate the GHR itself (termed an antagonistic action), rather it induces defective dimerization of the receptor, preventing subsequent signal transduction pathways.
Pegvisomant is thus a per can be considered a “true” antagonist of the GH-IGF1 axis. To date, pegvisomant is the most efficacious medical therapy for acromegaly, with responses seen in ~95% of patients. pegvisomant is generally well-tolerated, with the most common side effects being localized injection site reactions and flu-like symptoms. Rare but serious side effects include elevation of liver enzymes and the development of antibodies to pegvisomant. With long-term pegvisomant therapy, some patients may develop resistance.
Cabergoline is a dopamine agonist that has been shown to be effective in the treatment of growth hormone (GH)-secreting tumors. The mechanism by which cabergoline exerts its effects is not completely understood, but it is thought to work via the downregulation of D2 receptors. This is significant because GH-secreting tumors express D2R receptors to varying degrees, which determines their response to dopaminergic agonists. Indeed, the responsiveness of D2R’s on somatotroph tumors depends on their sensitivity and the concentration of circulating GH.
Cabergoline has been shown to be effective in the treatment of acromegaly, a condition characterized by excessive GH secretion. One advantage of cabergoline over other dopamine agonists is its long half-life, which allows for once-weekly dosing. Cabergoline is generally well-tolerated, with the most common side effects being nausea, vomiting, and headache.
| Medication | Mechanism of action | Dose |
| Somatostatin receptor ligands (Octreotide and lanreotide) | Affinity for somatostatin receptor subtypes 2 and 5. Suppresses the release of GH. | Octreotide (Initial dose is 20mg IM once monthly, increase to 30-40mg per month if IGF1 doses not normalize within 3 months). Lanreotide (30mg every 7-14days IM, 60-120mg q4-6 weeks as autogel or depot) |
| Somatostatin receptor ligands (Pasireotide) | Affinity for somatotropin receptor subtypes 1,2,3,5 (predominant affinity for subtype 5. Suppresses the release of GH. | Pasireotide (IM 40-60mg once monthly) |
| Dopamine receptor agonists | Dopamine, through its action on the hypothalamus, causes GH release by increasing GHRH and, through its action on the pituitary gland, inhibits GH release. D2 receptors are overexpressed on somatotropinoma, and DAs directly inhibit GH release through its action on the pituitary gland, thereby overcoming the hypothalamic effects. | Cabergoline (1-4mg) weekly (oral) |
| Pegvisomant | Growth hormone receptor antagonist | Pegvisomant (10-40mg) is administered SC once daily. |
Radiation Therapy
Stereotactic radiosurgery (SRS) is a type of radiation therapy that uses high doses of radiation to target small areas with great accuracy. This makes it an ideal treatment option for acromegaly, a condition characterized by the overproduction of growth hormone. SRS can be used to destroy the tumor without affecting surrounding healthy tissue. In many cases, it can lead to long-term remission and significantly improve quality of life. stereotactic radiosurgery is typically only used when other treatment options, such as surgery and medication, have failed.
What is the difference between SRS and conventional radiotherapy?
Conventional radiotherapy results in biochemical control in up to 35% of patients, within ten years of treatment. It, however, comes with a risk of local nerve injury, secondary brain tumors, and a not so negligible risk (2%) of a cerebrovascular accident.
Conversely, stereotactic radiosurgery provides more targeted therapy to the tumor, resulting in fewer complications.
Summary of the GH-IGF-1 axis
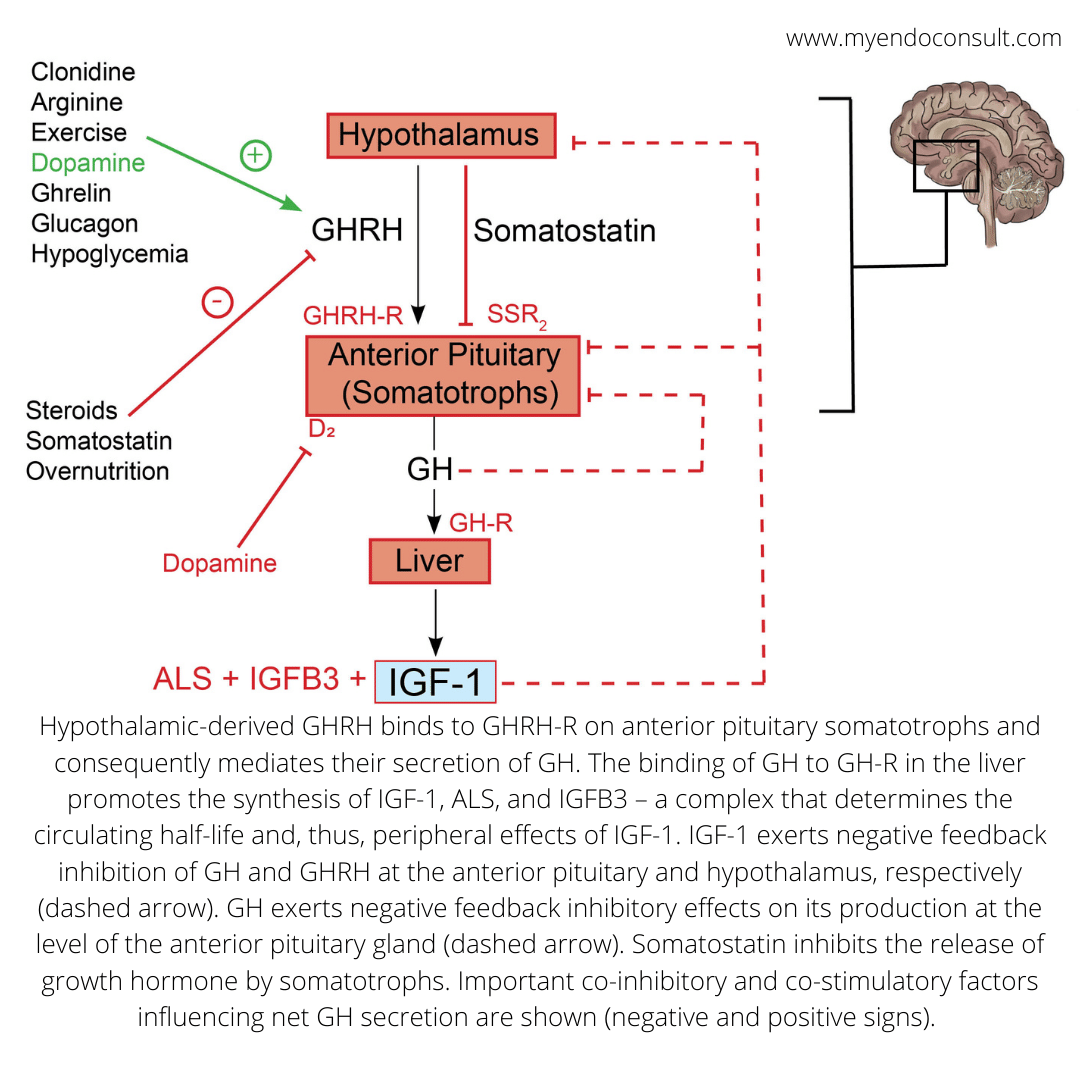 Summary of the GH-IGF-1 axis
Summary of the GH-IGF-1 axis
Prognosis
What is considered surgical amelioration (cure) of acromegaly?
- Normalization of age-adjusted IGF-1
- Restoration of growth hormone suppressibility below 0.4ng/dL on the 75g-OGTT
- Preservation of pituitary hormone function
- Complete removal of the tumor
What are the indications for long-term medical treatment of acromegaly?
- Multiple medical comorbidities or advanced aged and deemed a high surgical risk
- Invasive macroadenomas with parasellar extension (with no mass effects on vital structures)
- No discernible tumor, e.g., McCune-Albright syndrome (constitutive activation of the Gs alpha subunit)
Important clinical pearls
In normal pregnancy, growth hormone (GH) is nonsuppressible after an oral glucose load, and indeed, IGF-1 is elevated due to placental production of GH.
Thus the use of the standard OGTT is limited in the setting of pregnancy. Nonetheless, serum IGF-1 tends to be markedly elevated in pregnant women with acromegaly.
Interestingly, elevated levels of estrogen results in a relative GH resistant state which prevents the worsening of symptoms of acromegaly. Furthermore, GH and IGF-1 do not traverse the fetoplacental barrier, thus protecting the unborn baby from the deleterious effects of growth hormone excess.
If a pregnant woman with acromegaly develops symptoms suggestive of tumor expansion (visual field defects or worsening headaches), transsphenoidal surgery or medical therapy (SRL or DAs only) may be required.
References
- Ranke MB, Wit JM. Growth hormone – past, present and future. Nat Rev Endocrinol. 2018;14(5):285–300.
- Gariani K, Meyer P, Philippe J. Implications of Somatostatin Analogues in the Treatment of Acromegaly. Eur Endocrinol. 2013 Aug;9(2):132–5.
- Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM Int J Med. 2017 Jul 1;110(7):411–20.
- Giustina A, Barkhoudarian G et al. Multidisciplinary management of acromegaly: A consensus. Rev Endocr Metab Disord. 2020 Dec;21(4):667-678.
- Ershadinia N, Tritos NA. Diagnosis and Treatment of Acromegaly: An Update. Mayo Clin Proc. 2022 Feb;97(2):333-346.
- Holdaway IM. Treatment of acromegaly. Horm Res. 2004;62 Suppl 3:79-92.
Images(s) Courtesy
Case of Acromegaly WikiCommons