This is a rare X-linked genetic disorder of sex development characterized by mutations in the androgen receptor gene. A genetic male (XY genotype) will thus have tissues that are poorly sensitive to the effects of androgens. In effect, a male will have either a partial or full female phenotype[1].
Clinical features of Androgen Insensitivity Syndrome
Abnormalities of the external and internal genitalia
Patients with complete androgen insensitivity have a short vagina with an absent cervix on pelvic examination[2]. Due to the normal-appearing female external genitalia, the diagnosis is often delayed[3].
Normal breast tissue development
Patients with CAIS usually have normal female breast tissue development[4]. Aberrant breast tissue anywhere along the mammary line have been reported as well[5]. There is spontaneous breast development around puberty; as such, patients can grow up with a female gender identity[6]. The standard practice is to bring up CAIS subjects as females, although there is a recent case of gender dysphoria in a 17-year-old patient with CAIS. The patient underwent female-to-male gender reassignment surgery and subsequently received gender-affirming hormone therapy[7]. Aromatization of androgens into estrogen promotes breast tissue development in patients with CAIS[8].
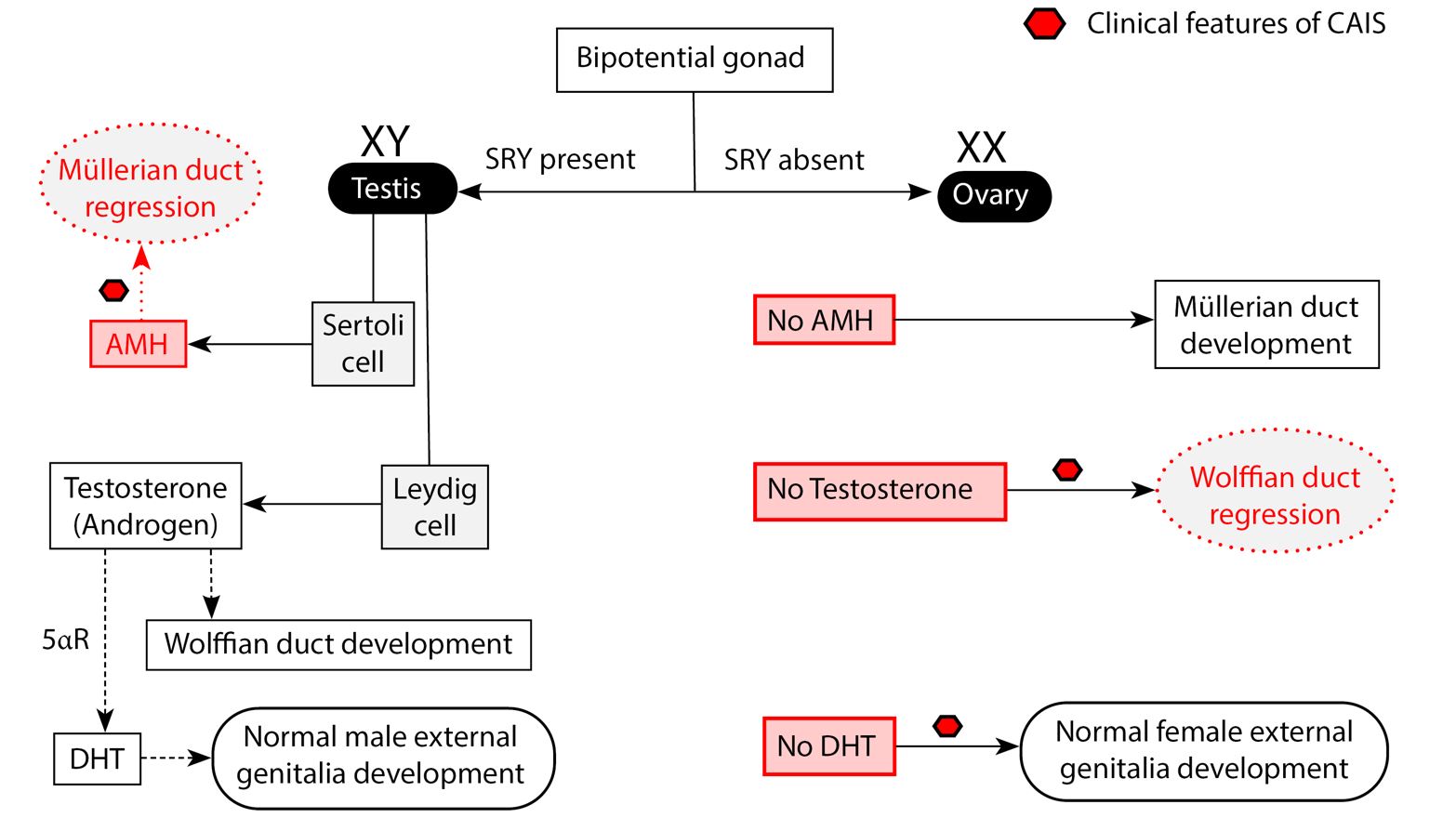
Fig. 1 Schematic diagram of normal differentiation of the bipotential gonad and the implications of complete androgen insensitivity. Complete androgen insensitivity syndrome (CAIS) occurs as a result of a mutation of the gene responsible for the translation of the androgen receptor (AR)[8, 9]. In utero, the bipotential gonad differentiates into either male or female gonad with ultimate phenotypic features being under the influence of the transcription factor, SRY (Sex-determining region of the Y chromosome), androgens and anti-Mullerian hormone (AMH) [4]. In a genetic male (XY), the SRY transcription factor present on the short arm of the Y chromosome plays a pivotal role in the differentiation of the bipotential gonad into a testis. AMH secreted by the Sertoli cells of the testis causes regression of the Mullerian ducts, which are responsible for the formation of the upper female genital structures (upper 1/3rd of the vagina, uterus and fallopian tubes)[4]. Androgens from the testes bind to androgen receptors and mediate the development of the Wolffian duct into male internal genital structures (seminal vesicles, epididymis, and vas deferens). Due to the mutation of the AR, the Wolffian ducts regress as well, leading to the formation of the lower female genitalia (lower 2/3rd vagina, labia, and clitoris)[4]. SRY = Sex-determining region of the Y chromosome, 5ɑR = 5 alpha-reductase (Redrawn and modified from Hughes et al. (2012) Androgen insensitivity syndrome. Lancet 380:1419–1428)
Clinical Pearls
Why should patients with CAIS and undescended testes undergo orchiectomy?
The risk of malignancy of an abdominally located testes increases with age, with an anticipated risk of malignancy of 3.6% at 25 years and an even higher risk of 33% by 50 years of age[10].
Why do patients with CAIS have incomplete or minimal axillary and pubic hair?
Vellus (nonpigmented or nonsexual) hair may be found in the pubic and axillary areas because it is not androgen-dependent. Darker terminal hair, which happens to be androgen-dependent, is, however, sparse or absent in CAIS. The inability of androgens to act at the hair root due to mutation of the androgen receptor accounts for these findings[11].
What are the other clinical features of CAIS?
Primary amenorrhea, inguinal hernias, sparse or absent pubic and axillary hair
Visual Abstracts
Disseminating scientific research in an engaging and easily digestible wayClick Here
References
- Farah S, El Masri D, Hirbli K (2021) Complete androgen insensitivity syndrome in a 13-year-old Lebanese child, reared as female, with bilateral inguinal hernia: a case report. Journal of Medical Case Reports 15:202
- Yang P, Liu X, Gao J, Qu S, Zhang M (2018) Complete androgen insensitivity syndrome in a young woman with metabolic disorder and diabetes: A case report. Medicine (Baltimore) 97:e11353
- Batista RL, Costa EMF, Rodrigues A de S, et al (2018) Androgen insensitivity syndrome: a review. Archives of Endocrinology and Metabolism 62:227–235
- Hughes IA, Davies JD, Bunch TI, Pasterski V, Mastroyannopoulou K, MacDougall J (2012) Androgen insensitivity syndrome. Lancet 380:1419–1428
- Nazzaro G, Genovese G, Brena M, Passoni E, Tadini G (2018) Aberrant breast tissue in complete androgen insensitivity syndrome. Clinical and Experimental Dermatology 43:491–493
- Tadokoro-Cuccaro R, Hughes IA (2014) Androgen insensitivity syndrome. Current Opinion in Endocrinology, Diabetes and Obesity 21:499
- T’Sjoen G, De Cuypere G, Monstrey S, Hoebeke P, Freedman FK, Appari M, Holterhus P-M, Van Borsel J, Cools M (2011) Male gender identity in complete androgen insensitivity syndrome. Arch Sex Behav 40:635–638
- Mongan NP, Tadokoro-Cuccaro R, Bunch T, Hughes IA (2015) Androgen insensitivity syndrome. Best Pract Res Clin Endocrinol Metab 29:569–580
- Mendoza N, Motos MA (2013) Androgen insensitivity syndrome. Gynecol Endocrinol 29:1–5
- Souhail R, Amine S, Nadia A, Tarik K, Khalid EK, Abdellatif K, Ahmed IA (2016) Complete androgen insensitivity syndrome or testicular feminization: review of literature based on a case report. Pan Afr Med J. https://doi.org/10.11604/pamj.2016.25.199.10758
- Quigley CA, de Bellis A, Marschke KB, El-Awady MK, Wilson EM, French FS (1995) Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocr Rev 16:271–321