The renin-angiotensin-aldosterone system (RAAS) is a complex endocrine system that controls blood pressure, fluid balance, and sodium retention. Furthermore, the RAAS has various effects on multiple organs via paracrine processes. This complex cascade of various enzymatic processes depends on an important rate-limiting enzyme known as renin. The factors involved in positive and negative feedback regulation of the RAAS, target effects of aldosterone and perturbations in the system that can result in endocrine disease will be reviewed next.
History of the discovery of the RAAS
Robert Tigerstedt, a physiologist from Finland, was the first to describe the vasopressor effects of the kidney extract he appropriately named renin in 1898. He was able to demonstrate that the injection of a kidney extract into the jugular veins of experimental rabbits resulted in a significant elevation in blood pressure. His discovery of renin went largely neglected until almost three decades later, when his results were replicated in a similar experiment using canines.
An overview of the RAAS cascade
In summary, the renin-angiotensin system is a cascade of related proteins and peptides (i.e., renin, angiotensinogen, angiotensin-converting enzyme, and angiotensins), which culminates in the formation of an active hormone known as angiotensin II. Angiotensin II exerts various target organ effects directly or via the stimulation of the adrenal hormone, aldosterone. Aldosterone subsequently promotes renal potassium wasting, renal sodium conservation, and water retention.
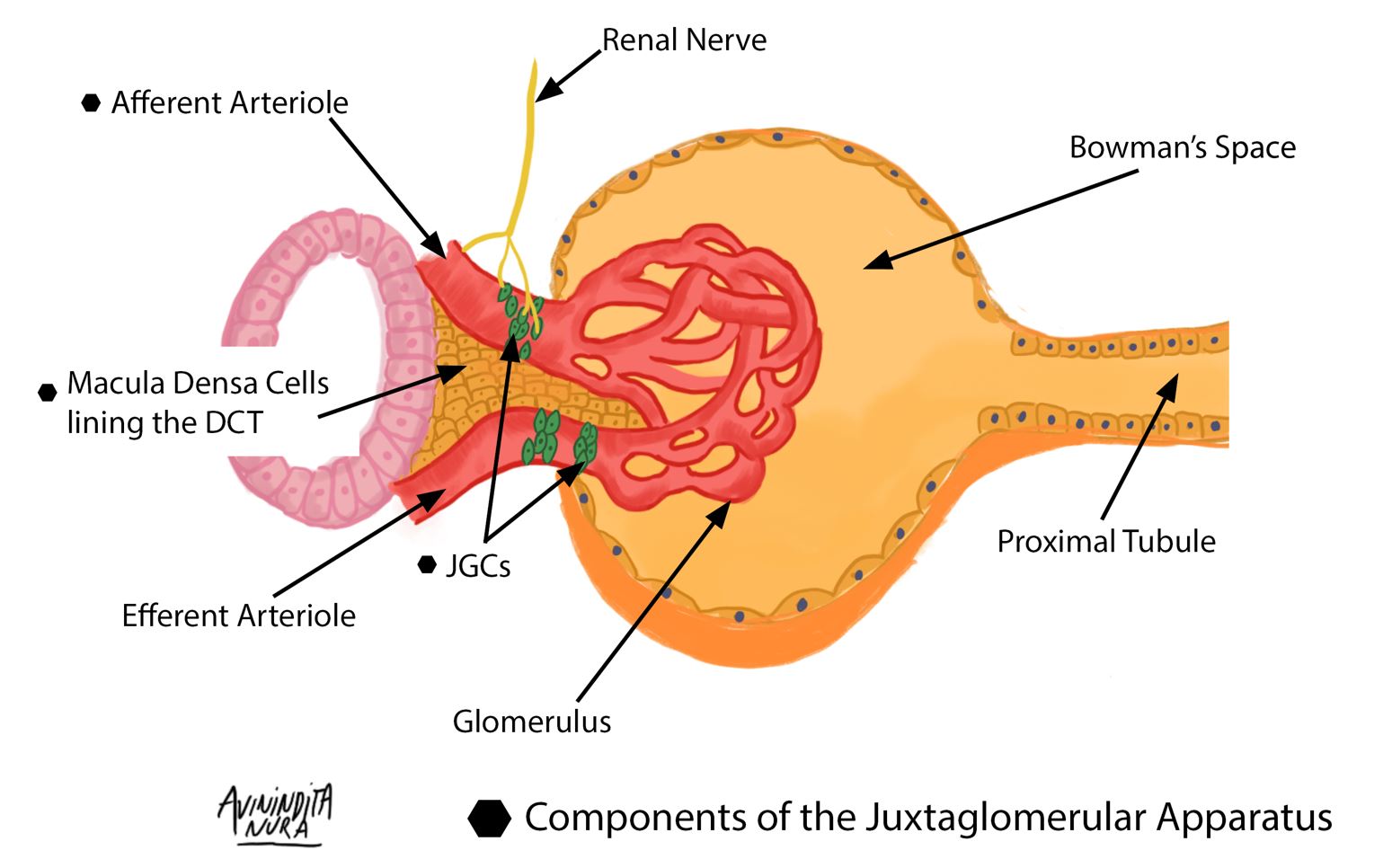
Triggers of renin secretion include a low sodium load presented to macula densa cells (located in the DCT), adrenergic stimulation from renal nerves innervating JGCs in the afferent arteriole, and a reduced tensile stretch of the afferent arteriole of the glomerulus. Renin, the rate-limiting enzyme in the renin-angiotensin-aldosterone system (RAAS), is critical in maintaining intravascular volume and blood pressure. Renin release is also controlled by long and short negative feedback loops mediated by the effect of ATII on the collecting tubule and JGCs, respectively. (Redrawn and modified from Alessandro et al. (2012) Role of the Renin-Angiotensin-Aldosterone System in the Pathogenesis of Atherosclerosis. Curr Pharm Des 18:981–1004)
A review of the juxtaglomerular apparatus is available here.
Renin – a critical rate-limiting enzyme
Renin is derived from preprorenin, a precursor molecule that is further cleaved and processed into prorenin, an inactive molecule. The majority of circulating renin exists in its inactive form with a ratio of 9:1, comparing prorenin to renin, respectively.
During embryonic development, renin is produced primarily by smooth muscle cells in intrarenal arterioles; however, the site of production is eventually restricted to the juxtaglomerular apparatus. It is worthy to note that various tissues, including those in the heart (smooth muscle cells in the vasculature), may be induced to produce renin when chronically stimulated. For example, a deficient sodium diet or exposure to ACE inhibitors can lead to this unique phenomenon known as “recruitment.”
Synthesis and secretion of the rate-limiting enzyme of this cascade herald various downstream processes critical in maintaining fluid and electrolyte balance. The secretion of renin occurs in response to various stimuli that result in target organ effects within a few minutes.
There are various pathways that regulate the secretion of renin.
· The intra-renal baroreceptor pathway: The universally accepted “baroreceptor pathway of renin secretion” was first proposed by Skinner and posits that increases or decreases in the tensile stretch (blood pressure) of afferent arterioles can either inhibit or stimulate the release of renin, respectively. A significant decline in blood pressure, mainly at the level of pre-glomerular (afferent arteriole) vessels, stimulates the renin release by the juxta glomerular cells.
· The macula densa pathway: The macula densa is a group of specialized cells present in the lining of the early distal tubule, which is in direct contact with the afferent and efferent arterioles of the glomerulus. The macula densa cells are sensitive to the sodium load present in the early distal tubule. For example, when there is reduced urinary sodium excretion, the macula densa cells transmit various complex paracrine chemical signals to nearby juxtaglomerular cells to induce the secretion of renin by the latter. Conversely, a decline in sodium delivery to the early distal tubule blunts the release of renin. Furthermore, prostaglandins, nitric oxide, and adenosine can stimulate the macula densa pathway.
· The beta-adrenergic receptor pathway: Catecholamines released directly from the adrenal medulla or renal sympathetic nerve endings (which innervate JGCs in the afferent arteriole) also stimulate renin release. The activation of beta 1 adrenergic receptors present on JGCs promotes renin release.
Angiotensinogen – A substrate for the enzyme renin
Angiotensinogen belongs to the alpha globulin fraction of plasma proteins and is synthesized primarily by the liver. Renin converts angiotensinogen to angiotensin I (an inactive decapeptide). The effects of renin on its substrate, i.e., angiotensinogen, are further augmented by various hormones, including cortisol, thyroid hormone, estrogens, and angiotensin II.
Angiotensin-Converting Enzyme (ACE)
ACE is a ubiquitous enzyme expressed in the vascular endothelium of various tissues found in the brain, skeletal muscle, skin, heart, kidney and lungs. This peptidase is abundant in the endothelium of the pulmonary vasculature and is required to convert angiotensin I to a potent octapeptide known as angiotensin II. As was previously mentioned, angiotensin II is a hormone that exerts various effects in distant tissues.
Furthermore, angiotensin converting enzyme 2 (ACE2) is required for negative feedback regulation of the RAAS. ACE2 converts angiotensin I, a decapeptide, to angiotensin (1-9), which undergoes further cleavage by ACE to a truncated heptapeptide known as angiotensin (1-7). Angiotensin (1-7), by binding to angiotensin type 2 (AT2) receptors, exerts a myriad of effects, including peripheral vasodilation and inhibition of anti-diuretic hormone release. This, in essence, counterbalances the fluid retentive and vasoconstrictive effects of angiotensin II.
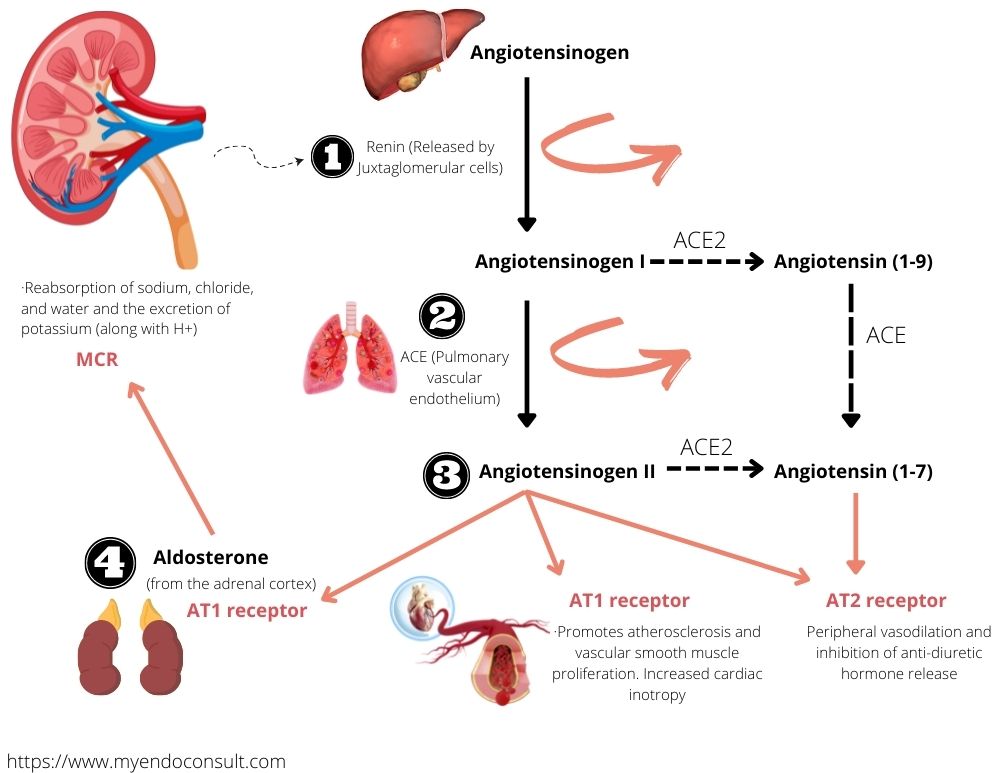
Schematic representation of the renin angiotensin aldosterone system RAAS.
Angiotensin II – a hormone with variable effects
Angiotensin II exerts various effects by binding to mainly two receptor subtypes that are angiotensin receptor type 1 (AT1) and AT2 receptors.
AT1 receptor is part of the G protein-coupled receptor family and s expressed in vessels, liver, lung, kidney, adrenal gland, and integument. The effects of the activation of AT1 receptors by angiotensin II include:
· Vasoconstriction
· Increased inotropic effects on cardiac muscle
· Potentiation of catecholamine release from sympathetic nerve endings
· Stimulation of aldosterone release by the zona glomerulosa of the adrenal cortex.
· Cardiac muscle proliferation
· Promotes atherosclerosis and vascular smooth muscle proliferation
· Stimulates phospholipase A2 in the arachidonic acid pathway. This promotes the release of various mediators required for renal and vascular regulation of systemic blood pressure.
· Increased activity of NADPH oxidase required in the generation of various reactive oxygen species.
The AT2 receptor is widely expressed in various organs, including the brain, kidneys, heart, adrenals, and integument. In contrast to the effects of AT1 receptor activation by angiotensin II, stimulation of the AT2 receptor counteracts the effects of the former.
The effects of AT2 receptor activation include:
· Inhibition of sympathetic activation
· Inhibition of inflammatory effects of AT1 receptor activation
· Vasodilation and urinary sodium loss
· Antithrombotic and Antifibrotic effects
Effects of aldosterone
Aldosterone is a mineralocorticoid produced by the zona glomerulosa of the adrenal cortex. A summary of the effects of aldosterone includes:
· Reabsorption of sodium, chloride, and water and the excretion of potassium (along with H+)
· In response to the sodium retentive effects of aldosterone, hypothalamic osmoreceptors detect this increase in serum osmolarity. Consequently, this stimulates the secretion of vasopressin (antidiuretic hormone) from the posterior pituitary gland, which then potentiates free water reabsorption in the collecting duct. An increase in extracellular volume dilutes the sodium pool, resulting in a reduction in plasma osmolarity.
· Vasopressin released in response to aldosterone-mediated sodium retention also exerts vasopressor effects in peripheral tissues.
· Aldosterone directly increases vascular tone by potentiating the vasopressor effects of catecholamines.
· Aldosterone also limits the effects of nitric oxide (a potent vasodilator).
· Aldosterone exerts various deleterious effects, including promoting vascular hypertrophy, inflammation, and oxidative stress.
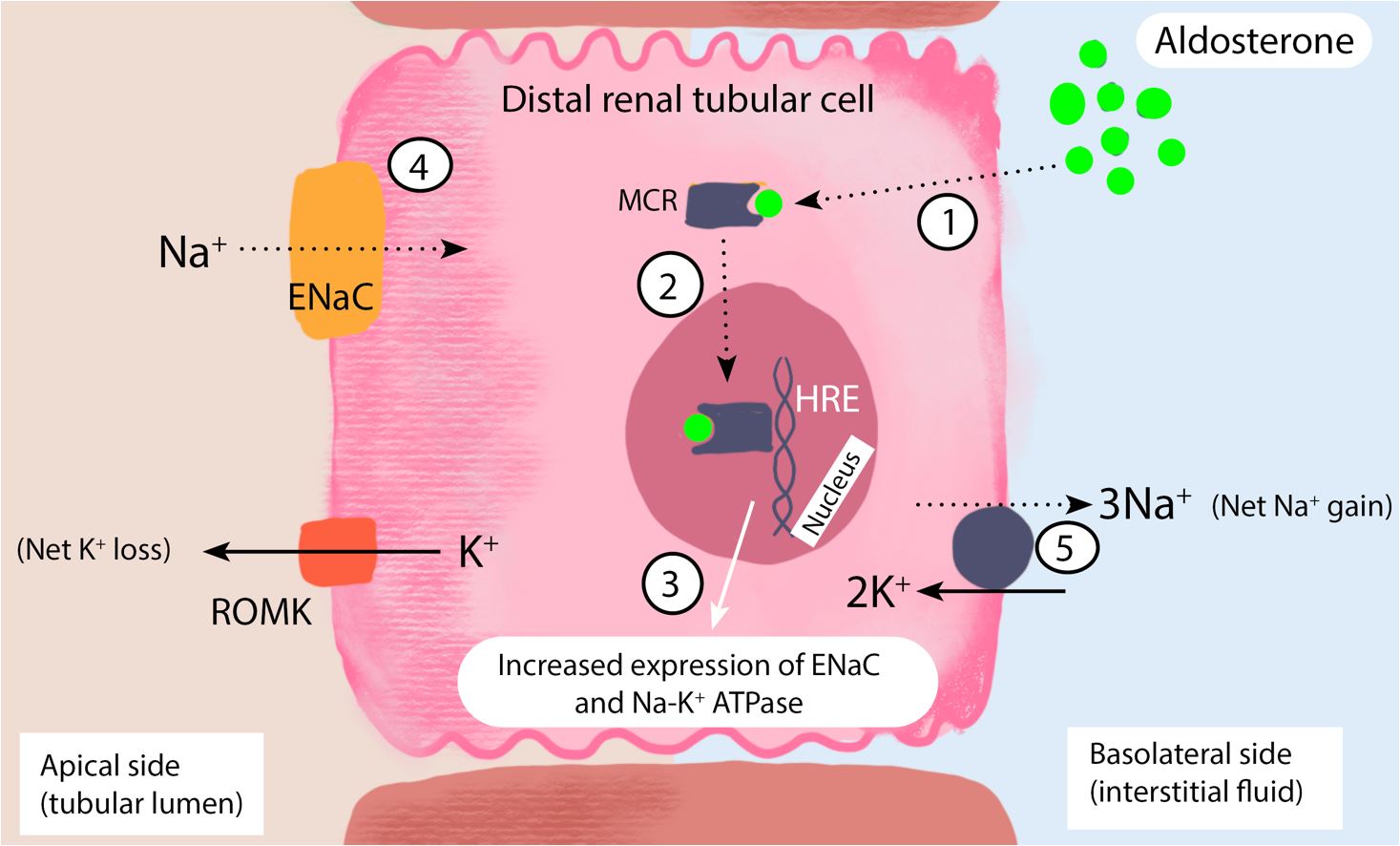
Aldosterone-mediated sodium and water conservation at the distal renal tubule.
Aldosterone, a lipid-soluble steroid hormone, diffuses through the cell membrane of the ductal epithelial cell and binds to the cytosolic mineralocorticoid receptor (MCR) (step 1). The aldosterone-MCR complex is then translocated into the nucleus, where it attaches to the hormone response element (HRE) (step 2) required for transcription and translation of the epithelial sodium chloride (ENaC) channel and sodium-potassium adenosine triphosphatase (Na-K ATPase) pump (step 3). ENaC and Na-K+ ATPase both play an active role in sodium reabsorption from the filtered renal sodium load present in the collecting duct and distal convoluted tubule. The net effect is the transfer of sodium from the apical to the basolateral side of the renal tubular cell (steps 4 and 5). Aldosterone also promotes the expression of renal outer medullary potassium (ROMK) channels. The insertion of ROMK channels on the apical membrane facilitates potassium and hydrogen ion loss.
Pathophysiology pearls
Why do patients with primary hyperaldosteronism seldom develop peripheral edema despite the sodium and water-conserving effects of excess aldosterone?
An increase in sodium and water conservation elevates the cardiac preload and leads to stretching of the atrial cardiomyocytes. Atrial natriuretic factor is then released by the atrial cardiomyocytes, which promotes natriuresis (renal sodium loss) in the medullary collecting ducts of the nephron.
Increased renal perfusion pressures due to plasma volume expansion lead to “pressure natriuresis.” The proximal tubule is unable to reabsorb enough sodium due to an increased filtered load in the setting of increased intraglomerular filtration pressures. This results in the delivery of a high renal tubular sodium load to the distal nephrons, which subsequently overwhelms the sodium-conserving effects of aldosterone. Learn more about aldosterone escape here.
How does hyperaldosteronism lead to atrial fibrillation?
1. Firstly, mineralocorticoid receptors present on cardiac myocytes are activated in the setting of hyperaldosteronism, which consequently results in myocyte hypertrophy. Furthermore, Left ventricular hypertrophy (LVH), which develops in response to myocyte hypertrophy, causes significant diastolic dysfunction and predisposes patients to atrial fibrillation.
2. Aldosterone exerts additional deleterious effects, including myocardial inflammation and fibrosis. Multiple foci of fibrosis scar cardiomyocytes and introduce potential re-entry circuits in the heart, which increases the risk of atrial fibrillation.
3. Hyperaldosteronism-induced hypokalemia causes prolongation of the PR interval (extends the period of ventricular diastolic filling), which impairs diastolic function and predisposes the myocardium to arrhythmias.
References
1. Byrd James Brian, Turcu Adina F., Auchus Richard J. Primary Aldosteronism. Circulation. 2018;138(8):823-835.
2. Ong GSY, Young MJ. Mineralocorticoid regulation of cell function: the role of rapid signalling and gene transcription pathways. J Mol Endocrinol. 2017;58(1):R33-R57. doi:10.1530/JME-15-0318
3. Valinsky WC, Touyz RM, Shrier A. Aldosterone, SGK1, and ion channels in the kidney. Clin Sci Lond Engl 1979. 2018;132(2):173-183. doi:10.1042/CS20171525
4. Schrier RW. Aldosterone “escape” vs “breakthrough.” Nat Rev Nephrol. 2010;6(2):61. doi:10.1038/nrneph.2009.228
5. Seccia TM, Caroccia B, Adler G, Maiolino G, Cesari M, Rossi GP. Arterial Hypertension, Atrial Fibrillation And Hyperaldosteronism: The Triple Trouble. Hypertens Dallas Tex 1979. 2017;69(4):545-550.
6. Learn more endocrine pathophysiology here [Endocrine Pathophysiology – A Concise Guide to the Physical Exam]