This is a concise review of AHO, including its initial description by Fuller Albright, classic clinical features, pathophysiology, and genetic background.
What is the classic phenotype of AHO?
Dr. Fuller Albright first described Albright’s hereditary osteodystrophy (AHO) in 1942. The classic phenotype has the following features, brachydactyly, short stature, and round facies. The classification system for this group of inactivating PTH/PTHrp signaling disorders is based on the presence or absence of the classic AHO phenotype and Gsɑ activity in response to exogenous PTH administration[1].
Short Stature
Short stature is a cardinal clinical finding in Albright hereditary osteodystrophy (AHO). The classic AHO phenotype is characterized by round facies, obesity, brachydactyly, and short stature[2]. Short stature in patients with pseudohypoparathyroidism (PHP) occurs due to rapid chondrocyte differentiation leading to premature closure of the growth plate and eventual stunting of growth[3]. Impaired activity of Gsɑ affects PTH mediated signaling in chondrocytes. This is even more profound during puberty and accounts for the short stature observed in patients with pseudohypoparathyroidism[4].
Obesity
Most adults with PHP1A have a body mass index (BMI) >25kg/m2[4]. The reported prevalence of obesity is more than 66%, higher than the 32% prevalence rate reported for the general population[5].
- Melanocortin signaling pathways that regulate satiety are dependent on Gsɑ activity. Gsɑ activity is defective in PHP, which leads to impaired satiety, ultimately leading to obesity[4].
- There are β adrenergic receptors in adipose tissue, which presumably play a role in lipolysis. Downstream signaling of these receptors is dependent on Gsɑ activity; as such, decreased fat mobilization occurs in patients with PHP[6].
- Growth hormone (GH) deficiency is a contributory factor as well since Growth hormone-releasing hormone (GHRH) signaling is dependent on the stimulatory G-protein, Gsɑ[5].
Brachydactyly
The characteristic skeletal feature of PHP happens to be shortening of the metacarpals and metatarsals. This skeletal change tends to involve the fourth and fifth metacarpals and metatarsals [4, 7]. Impaired activity of Gsɑ affects Parathyroid hormone-related peptide (PTHrp) mediated signaling critical in chondrocyte proliferation in the growth plate[4].
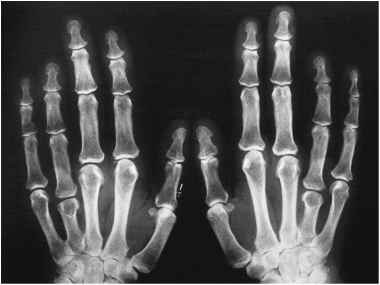
Figure 1. Plain radiograph of both hands depicting brachydactyly – shortening of the 4th metacarpals bilaterally.
Dental manifestations.
Multiple dental abnormalities include delayed or even failed eruption of the teeth, blunting of the roots, hypodontia, and ankylosis[4, 8]. Patients should be referred to dentists for optimal care of their teeth[8]. Downstream signaling for PTH-1R in the tooth is affected due to impaired Gsɑ activity. This highlights the vital role of PTH in mediating tooth maturation and mineralization[4].
Pathophysiology Pearl
PTH binds to the PTH-1R receptor, which leads to the dissociation of Gsɑ from the heterotrimeric G protein. Gsɑ subsequently activates adenylyl cyclase (AC). This is followed by AC-mediated conversion of ATP to cyclic AMP. The second messenger, cyclic AMP activates protein kinase A (PKA), which subsequently phosphorylates various target proteins involved in the transcription and translation of several downstream cyclic AMP-responsive genes.
A loss of function mutation involving the gene encoding the alpha subunit of the Gs protein, i.e., the GNAS gene, results in pseudohypoparathyroidism[4]. Resistance to PTH action in the proximal renal tubule leads to hypocalcemia and hyperphosphatemia, a biochemical profile akin to what is observed in patients with isolated hypoparathyroidism. Patients, however, have a paradoxical elevation in serum PTH, hence the name, pseudohypoparathyroidism[4]
Pseudopseudohypoparathyroidism (PPHP). This is a unique clinical and biochemical subtype of inactivating PTH/PTHrp signaling disorders (iPPSD). The new iPPSD nomenclature acknowledges the spectrum of clinicopathologic phenotypes in patients with pseudohypoparathyroidism[1].
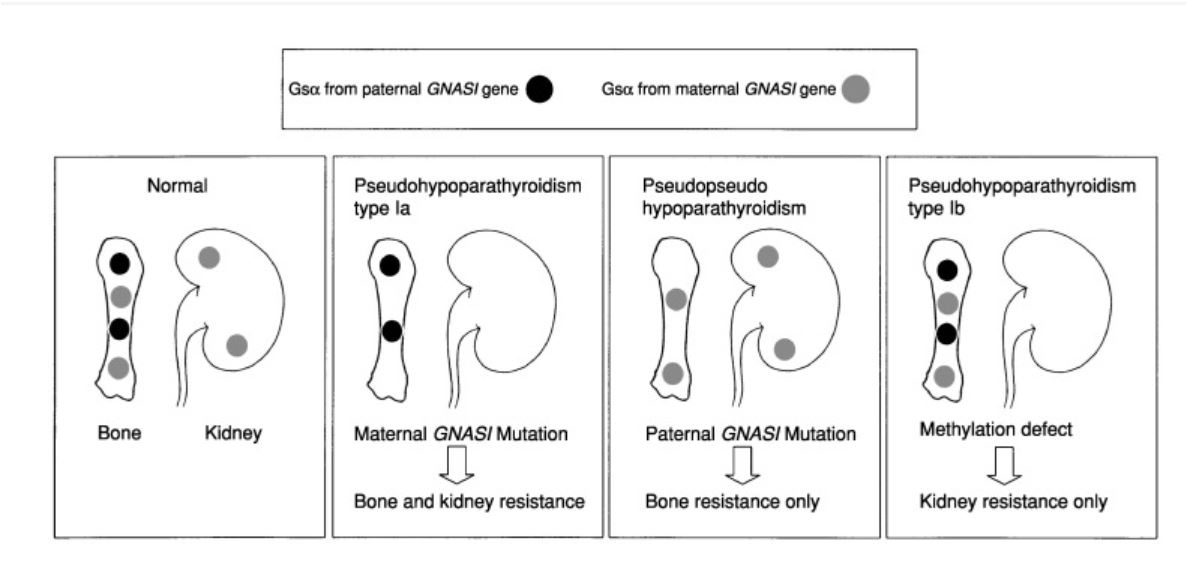
Figure 2. Role of tissue specific expression of Gsα gene in determining the final clinical phenotype of pseudohypoparathyroidism. The Guanine nucleotide-binding protein, alpha stimulating (GNAS) gene, is critical in the transcription of the stimulatory G protein (Gsα). Gsα in most tissues is expressed in a biallelic fashion, i.e., there are distinct paternal and maternal alleles. The clinical and biochemical features are, therefore, dependent on the parent of origin of the mutant allele[9].
The paternal Gsα gene in normal physiology is not expressed in the proximal renal tubule, pituitary gland, and gonadal tissue. It, therefore, plays no role in renal electrolyte (calcium and phosphorus) handling or activation of Gsα coupled receptors such as luteinizing hormone (LH), parathyroid hormone (PTH), and thyroid-stimulating hormone (TSH). Paternal derived Gsα gene is however present in bone. An affected child who inherits a mutated Gsα gene from a father will, therefore, present with pseudopseudohypoparathyroidism PPHP (i.e., short stature with no apparent biochemical or hormonal perturbations)[9].
Maternal Gsα gene expression determines most of the downstream effects of Gsα coupled receptors in extra-skeletal tissues, including LH, PTH, TSH, and GHRH. Also, unlike the paternal allele, the maternal allele is expressed in several tissues including pituitary, renal, and gonadal tissues. It is noteworthy, that both maternal and paternal Gsα genes are expressed in bone. A mutation in the maternal Gsα gene, therefore, results in the classic Pseudohypoparathyroidism type 1A (PHP1A) phenotype (see table 1)[10, 11].
Pseudohypoparathyroidism type 1B (PHP1B) occurs when there is an imprinting (methylation) defect in the maternal GNAS gene. However in contrast to pseudopseudohypoparathyroidism and PHP1A, there is no mutation in the Gsα gene[12].
| iPPSD | AHO | Other hormone Resistance States. | PTH resistance |
|---|---|---|---|
| PPHP | Present | Absent | Absent [13, 14] |
| PHP1A | Present | Present | Present [10, 11] |
| PHP1B | Absent | Infrequent | Present [12]. |
| PHP1C | Present | Present | Present [2]. |
- iPPSD inactivating PTH/PTHrp signaling disorders, AHO Albright’s hereditary osteodystrophy.
- Other hormone resistance states LH and TSH resistance
- PTH resistance low calcium, high phosphorus and a paradoxically high PTH
- PHP1C Pseudohypoparathyroidism type 1C
What are the other endocrinopathies which might be associated with some forms of pseudohypoparathyroidism?
- Resistance to the effects of TSH at the level of the thyroid gland results in hypothyroidism
- Gonadotropin resistance leading to delayed puberty, oligomenorrhea and cryptorchidism
- Growth hormone-releasing hormone (GHRH) resistance causes GH deficiency.
- Prolactin deficiency
Interestingly, ACTH, CRH, and vasopressin action are not affected in iPPSDs, because both the maternal and paternal copies of the GNAS gene are expressed in their target tissues, as such a mutation in one parental allele does not result in hormonal defects since the normal parental allele is present. This highlights the tissue-specific expression of both maternal and paternal copies of the gene[15].
References
- Turan S (2017) Current Nomenclature of Pseudohypoparathyroidism: Inactivating Parathyroid Hormone/Parathyroid Hormone-Related Protein Signaling Disorder. J Clin Res Pediatr Endocrinol 9:58–68
- Mantovani G, Bastepe M, Monk D, et al (2018) Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol 14:476–500
- Hanna P, Grybek V, Nanclares GP de, et al (2018) Genetic and Epigenetic Defects at the GNAS Locus Lead to Distinct Patterns of Skeletal Growth but Similar Early-Onset Obesity. Journal of Bone and Mineral Research 33:1480–1488
- Linglart A, Levine MA, Jüppner H (2018) Pseudohypoparathyroidism. Endocrinology and Metabolism Clinics of North America 47:865–888
- Long DN, McGuire S, Levine MA, Weinstein LS, Germain-Lee EL (2007) Body Mass Index Differences in Pseudohypoparathyroidism Type 1a Versus Pseudopseudohypoparathyroidism May Implicate Paternal Imprinting of Gαs in the Development of Human Obesity. J Clin Endocrinol Metab 92:1073–1079
- Carel JC, Le Stunff C, Condamine L, Mallet E, Chaussain JL, Adnot P, Garabédian M, Bougnères P (1999) Resistance to the Lipolytic Action of Epinephrine: A New Feature of Protein Gs Deficiency. J Clin Endocrinol Metab 84:4127–4131
- Linglart A, Fryssira H, Hiort O, et al (2012) PRKAR1A and PDE4D Mutations Cause Acrodysostosis but Two Distinct Syndromes with or without GPCR-Signaling Hormone Resistance. J Clin Endocrinol Metab 97:E2328–E2338
- Reis MTA, Matias DT, Faria MEJ de, Martin RM (2016) Failure of tooth eruption and brachydactyly in pseudohypoparathyroidism are not related to plasma parathyroid hormone-related protein levels. Bone 85:138–141
- Turan S, Bastepe M (2015) GNAS spectrum of disorders. Curr Osteoporos Rep 13:146–158
- Thiele S, Mantovani G, Barlier A, et al (2016) From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol 175:P1–P17
- Mantovani G, Elli FM (2019) Inactivating PTH/PTHrP Signaling Disorders. Parathyroid Disorders 51:147–159
- Dixit A, Chandler KE, Lever M, Poole RL, Bullman H, Mughal MZ, Steggall M, Suri M (2013) Pseudohypoparathyroidism Type 1b due to Paternal Uniparental Disomy of Chromosome 20q. J Clin Endocrinol Metab 98:E103–E108
- Elli FM, deSanctis L, Ceoloni B, Barbieri AM, Bordogna P, Beck-Peccoz P, Spada A, Mantovani G (2013) Pseudohypoparathyroidism type Ia and pseudo-pseudohypoparathyroidism: the growing spectrum of GNAS inactivating mutations. Hum Mutat 34:411–416
- Simpson C, Grove E, Houston BA (2015) Pseudopseudohypoparathyroidism. Lancet 385:1123
- Mantovani G (2011) Pseudohypoparathyroidism: Diagnosis and Treatment. J Clin Endocrinol Metab 96:3020–3030