

Aldosterone escape has been applied to two pathophysiologic states encountered in practice. The first is the classic “aldosterone escape” of primary hyperaldosteronism. Another form of aldosterone escape has been described among patients with heart failure treated with angiotensin-converting enzyme inhibitors.
The latter is somewhat of a misnomer and may be better termed aldosterone breakthrough and will not be discussed here. However, this review will focus on the classic “aldosterone escape” of primary hyperaldosteronism.
The renin-angiotensin-aldosterone system
The renin-angiotensin-aldosterone system is a hormone system that helps to regulate blood pressure. Renin is an enzyme that is produced by the kidney in response to low blood pressure or a reduction in blood flow. This enzyme triggers the release of angiotensin, a hormone that constricts blood vessels and increases blood pressure.
Once released, angiotensin triggers the release of aldosterone, another hormone that helps retain salt and water in the body. This process ultimately leads to an increase in blood volume and pressure, which helps to bring blood flow back to normal levels.
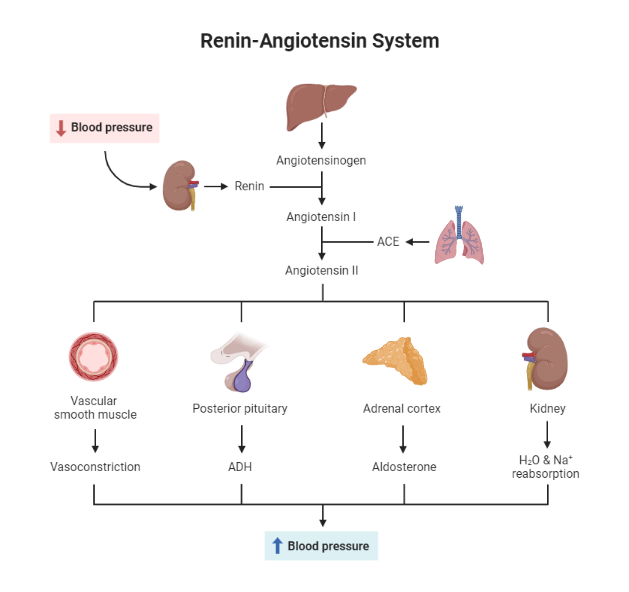
Fig 1. The Renin Angiotensin Aldosterone Axis
[tqb_quiz id=’4412681′]Aldosterone and water conservation
Aldosterone is a hormone that plays an essential role in the regulation of sodium and water balance in the body. The adrenal glands secrete it in response to blood pressure, electrolyte levels, and fluid status changes. Under normal circumstances, aldosterone helps conserve sodium and water by promoting reabsorption in the kidney.
This process, known as the renin-angiotensin-aldosterone system (RAAS), helps maintain blood volume and blood pressure within a normal range. In addition, aldosterone also plays a role in potassium homeostasis.
By promoting potassium excretion, aldosterone helps to maintain a healthy electrolyte balance in the body. Therefore, aldosterone is essential for the normal physiology of sodium and water conservation.
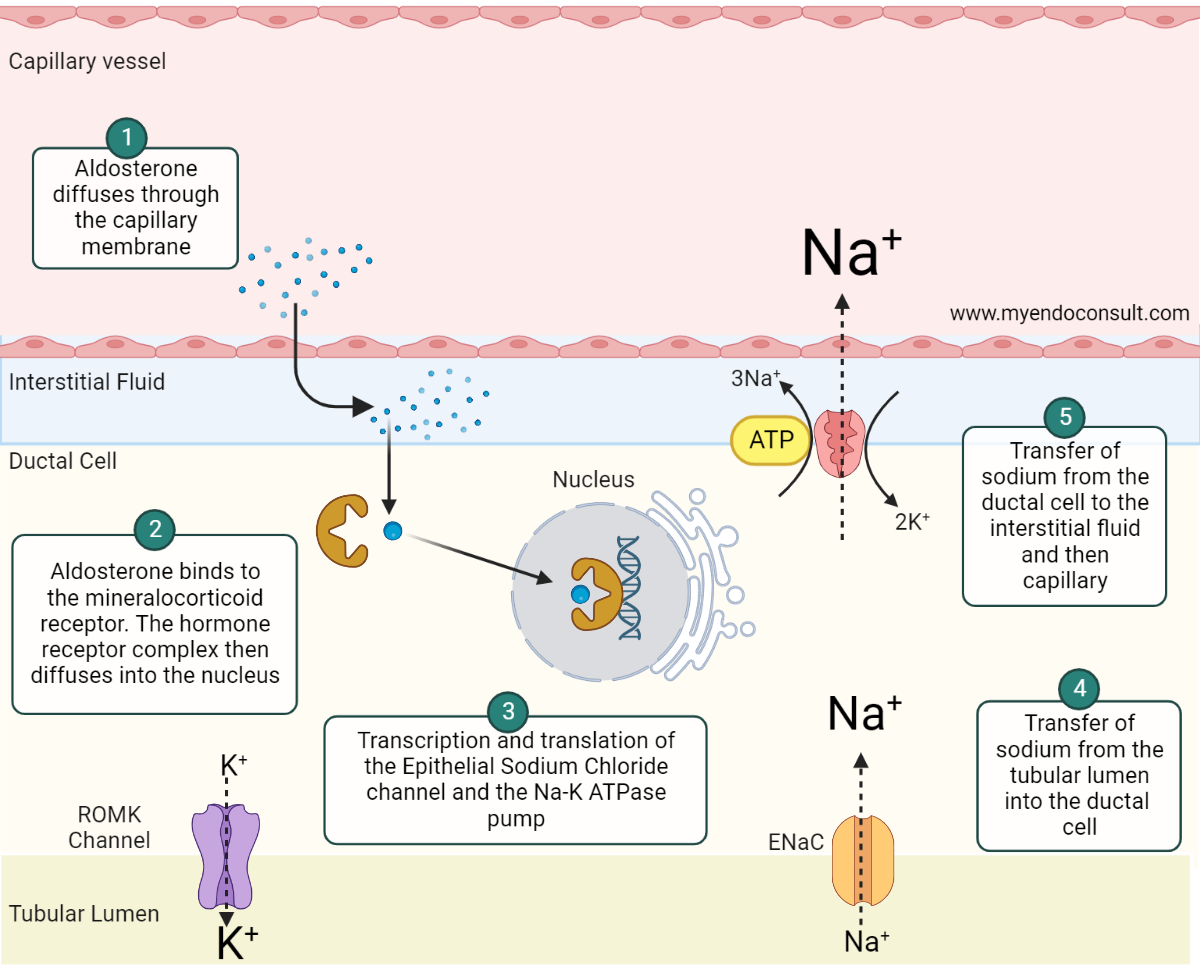
Fig 2.Aldosterone-mediated sodium and water conservation at the distal renal tubule.
Aldosterone, a lipid-soluble steroid hormone, diffuses through the cell membrane of the ductal epithelial cell and binds to the cytosolic mineralocorticoid receptor (MCR) (step 1). The aldosterone-MCR complex is then translocated into the nucleus where it attaches to the hormone response element (HRE) (step 2) required for transcription and translation of the epithelial sodium chloride (ENaC) channel and sodium-potassium adenosine triphosphatase (Na-K ATPase) pump (step 3). ENaC and Na-K+ ATPase both play an active role in sodium reabsorption from the filtered renal sodium load present in the collecting duct and distal convoluted tubule. The net effect is the transfer of sodium from the apical to the basolateral side of the renal tubular cell (steps 4 and 5). Aldosterone also promotes the expression of renal outer medullary potassium (ROMK) channels. The insertion of ROMK channels on the apical membrane facilitates potassium and hydrogen ion loss.
Previous Proposed Mechanisms which have been disproven.
| Site of Action | Mechanism |
| Mineralocorticoid Receptor | Reduced sensitivity of the mineralocorticoid receptor to aldosterone is an unlikely mechanism, because patients with primary hyperaldosteronism often have hypokalemia (also mediated in part by ENaC channels) |
| Downregulation of sodium channels including Na+-H+ antiporter in the proximal tubule or the Na+/K+/2Cl− symporter in the thick ascending loop of Henle | If downregulation of these sodium channels in the renal tubule were a possible mechanism of aldosterone escape, additional electrolyte perturbations would have been expected. |
Current Accepted Mechanism of Aldosterone Escape
 Fig 3. Aldosterone Escape Mechanism
Fig 3. Aldosterone Escape Mechanism
Hyperaldosteronism increases sodium and water reabsorption while promoting renal potassium and H+ losses. Consequently, hyperaldosteronism promotes plasma volume expansion, hypokalemia, and metabolic alkalosis. Plasma volume expansion increases renal blood flow (which decreases renin and angiotensin II production). Increased peritubular capillary hydrostatic pressure facilitates the flow of sodium from the interstitium back into the renal tubules (natriuresis). Also, review the role of Starling’s forces in regulating sodium handling at the renal tubules (see figure 3). In addition, stretching of atrial myocytes causes the excretion of atrial natriuretic factor (ANF). ANF then promotes the excretion of sodium despite hyperaldosteronism.
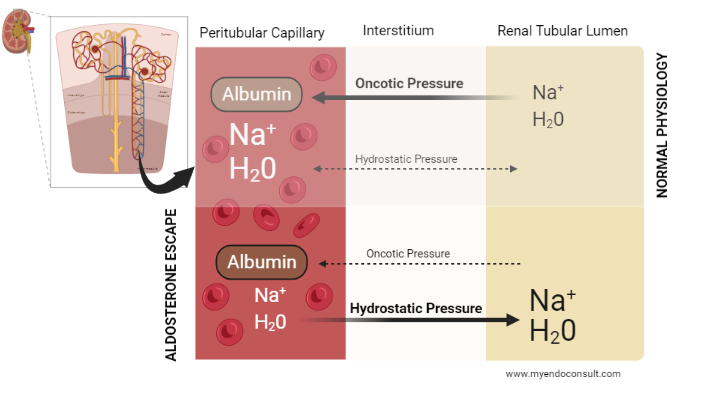
Fig 4. Role of Starling’s Forces in the pathophysiology of aldosterone escape
Under normal physiological conditions, sodium and water are reabsorbed from the renal tubular lumen and transported into the interstitium. Starling’s forces subsequently define the gradient of movement of sodium and water from the interstitium into the peritubular capillaries and, subsequently, the systemic circulation.
It is worth noting that hydrostatic pressures across the renal tubules, interstitium and peritubular capillaries are balanced in normal physiology. As a consequence, oncotic pressures dictate the flow of sodium and water. Because protein is much higher in the peritubular capillary compared to the interstitium, sodium and water are transported from the interstitium into the capillaries (via oncotic forces).
Conversely, in the setting of hyperaldosteronism, plasma volume expansion results in an overwhelmingly high hydrostatic pressure compared to the oncotic pressure in the peritubular capillaries. This pathologic shift in Starling’s forces promotes the “backflow” of sodium and water from the interstitium into the renal tubular lumen (pressure natriuresis). An adaptation that prevents hypernatremia and fluid retention (edema) in primary hyperaldosteronism.
References
Schrier, R. Aldosterone ‘escape’ vs ‘breakthrough’. Nat Rev Nephrol 6, 61 (2010).
Kindly Let Us Know If This Was helpful? Thank You!