Metformin, a pharmaceutical compound classified as a biguanide, demonstrates a multitargeted mechanism of action, effectively intervening in multiple pathophysiological processes implicated in type 2 diabetes mellitus.
• It mitigates hepatic gluconeogenesis, thereby limiting excessive endogenous glucose production
• Promotes glucose absorption in both skeletal muscle and adipose tissue, contributing to glucose homeostasis by reducing hyperglycemia.
• It improves insulin sensitivity, notably by upregulating intracellular insulin receptor substrate (IRS) signaling pathways, which play an integral role in insulin-mediated cellular functions.
Physiology
Glucose Metabolism
As the principal energy source for all cellular functions within the human body, plasma glucose levels must be meticulously maintained within a specific physiological range (64.8 to 104.4mg/dl), regardless of dietary intake. This balance is achieved through intricate and interlinked mechanisms designed to prevent both hypoglycemia and hyperglycemia.
During periods of fasting, the risk of hypoglycemia, or a significant decline in plasma glucose, is averted by adaptive processes such as gluconeogenesis, which involves the synthesis of glucose from noncarbohydrate sources, and glycogenolysis, the breakdown of glycogen stores.
Conversely, in the postprandial period, which is characterized by hyperglycemia, there is the activation of other processes: glycogenesis, the synthesis of glycogen from glucose, and the conversion of glucose to triacylglycerols, or fats.
The distribution and metabolism of plasma glucose among various tissues are highly dependent on the metabolic state, fasting or postprandial.
Gluconeogenesis
The human body typically requires an average of 160 grams of glucose per day, 75% of which is chiefly consumed by the central nervous system for energy production.
However, the readily accessible stores of glucose in humans are quite limited, with approximately 20 grams of glucose present in body fluids and roughly 190 grams stored as glycogen.
Depending on the energy demands of an individual, additional glucose sources may be necessary. In fact, without external dietary glucose intake, survival beyond 24 hours would be untenable, as most of these reserves would be exhausted. Therefore, gluconeogenesis, the endogenous production of glucose from non-glucose precursors such as pyruvate, lactate, glycerol, and amino acids, becomes a vital process. It predominantly occurs in hepatocytes and renal tissues, contributing 75% and 25% of glucose produced via gluconeogenesis, respectively.
Origins of Essential Glucose Precursors for Gluconeogenesis
Each precursor involved in gluconeogenesis is derived from distinct metabolic activities within the body:
- Lactate: This molecule originates from the anaerobic respiration process in skeletal muscles and subsequently undergoes conversion to pyruvate via the enzyme lactate dehydrogenase in the liver.
- Glycerol: This precursor arises from the hydrolysis of triglycerides within adipose tissues. Post-production, glycerol is transformed into dihydroxyacetone phosphate (DHAP), thus preparing it for its role in gluconeogenesis.
- Amino Acids: Generated through protein hydrolysis in dietary intake or skeletal muscle, amino acids are metabolized into either pyruvate or DHAP to further participate in gluconeogenesis.
The initial stages of gluconeogenesis, specifically the first two steps, aim to transform pyruvate into phosphoenolpyruvate (PEP), involving oxaloacetate as an intermediate. This conversion process requires the input of ATP and carbon dioxide.
Upon the formation of PEP, a sequence of enzymatic reactions (steps 3-7) culminates in the production of fructose 1,6-bisphosphate. Interestingly, these steps essentially mirror the inverse reactions that occur during glycolysis.
Under the regulation of the allosteric enzyme, fructose 1,6 bisphosphatase, fructose 1,6-bisphosphate undergoes hydrolysis to yield fructose 6-phosphate. This compound is then subject to isomerization, catalyzed by phosphoglucose isomerase, to generate glucose-6-phosphate. The metabolic pathway of glucose-6-phosphate diverges substantially, depending on the tissue in which it is formed.
Notably, glucose-6-phosphate is unable to cross cell membranes, effectively making it a readily available substrate for subsequent metabolic processes. For instance, within skeletal muscle, glucose-6-phosphate can either be stored as glycogen or be metabolized into pyruvate for energy production through glycolysis .
On the other hand, in hepatocytes and renal cells, glucose-6-phosphate is transformed into glucose via the enzyme glucose-6-phosphatase. Owing to the presence of glucose transporters on cellular membranes, this glucose can readily exit these principal sites of gluconeogenesis and be transported to other cells throughout the body.
Glycolysis
Glycolysis represents a series of biochemical reactions that effectively convert a glucose molecule into two molecules of pyruvate, two molecules of ATP, two molecules of NADH, and two molecules of water.
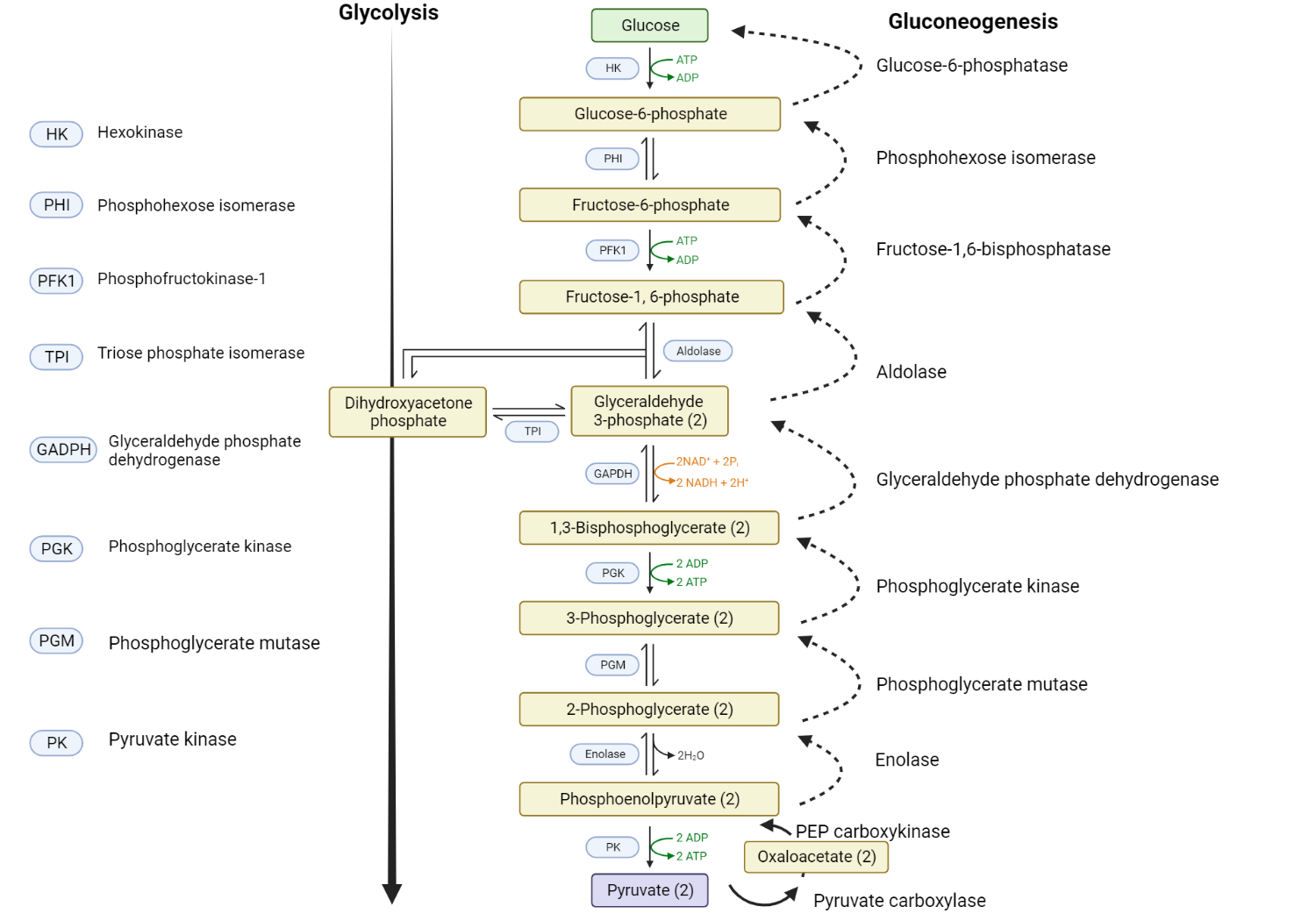
The initial step in glycolysis involves the conversion of glucose to glucose-6-phosphate by the action of the enzyme hexokinase. This conversion destabilizes the glucose molecule and, crucially, traps it within the cell. Subsequently, the enzyme phosphoglucose isomerase mediates the isomerization of glucose-6-phosphate into fructose-6-phosphate. The enzyme phosphofructokinase then facilitates the phosphorylation of fructose-6-phosphate, transforming it into fructose-1,6-bisphosphate. Following these steps, a series of enzymatic reactions ensue, culminating in the formation of pyruvate.
Glycogenesis
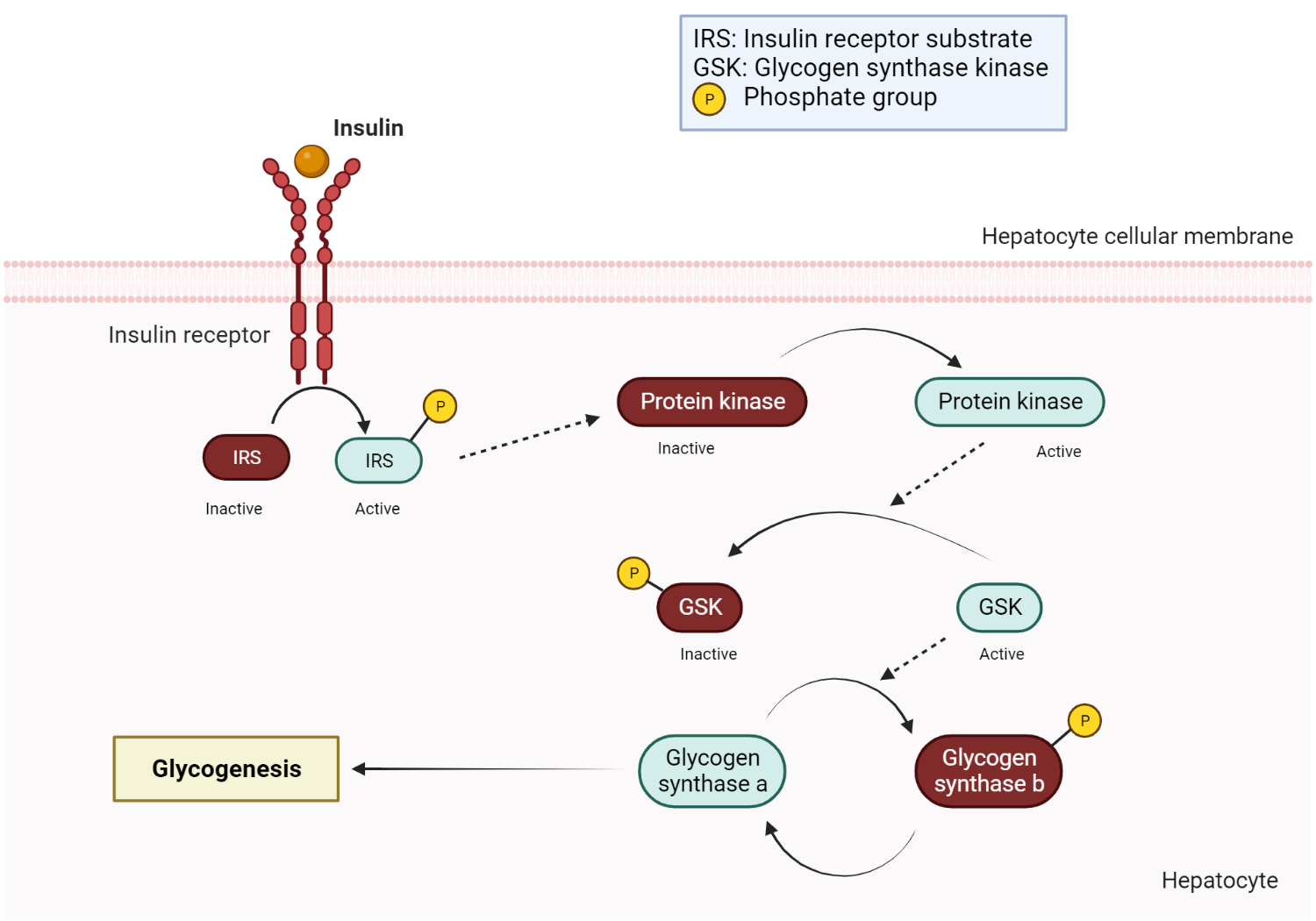
Glycogenesis denotes the process of glycogen synthesis, whereby glycogen, a stored form of glucose, is produced. Upon consuming a carbohydrate-rich meal, blood glucose levels rise, a change detected by the pancreatic beta cells, which subsequently secrete insulin. The insulin hormone then circulates to hepatocytes, where it binds to insulin receptors and triggers a signal transduction pathway activating various cytoplasmic protein kinases.
These activated protein kinases serve to inactivate glycogen synthase kinase, an enzyme typically responsible for the inactivation of glycogen synthase. As a result, glycogen synthesis is facilitated due to the reduction in inhibitory control over the central regulatory enzyme, glycogen synthase.
In the context of insulin binding to its receptor, a signaling cascade is initiated, which involves the activation of insulin receptor substrate (IRS) and phosphatidylinositol 3-kinase (PI3K). This sequence of events leads to the activation of Akt (protein kinase B), which inhibits glycogen synthase kinase-3 (GSK-3). By inhibiting GSK-3, the conversion of inactive glycogen synthase b to its active form, glycogen synthase a, is promoted, thereby facilitating glycogen synthesis and storage. Ultimately, glycogen synthase catalyzes the elongation of glycogen by sequentially attaching glucose monomers via alpha 1,4 glycosidic bonds.
Glycogenolysis
Glycogenolysis refers to the process of glycogen breakdown or degradation, which involves a sequence of enzymatic actions, culminating in the release of glucose from glycogen. The process begins with an initial phosphorolysis step, requiring the enzyme glycogen phosphorylase. This is followed by a glycogen remodeling phase, involving a transferase and alpha 1,6 -glucosidase enzymes. The remodeled glycogen molecule then releases glucose, which is converted by the hexokinase enzyme into glucose-1-phosphate. Subsequently, phosphoglucomutase acts on glucose-1-phosphate, transforming it into glucose-6-phosphate.
As previously mentioned, the metabolic fate of glucose-6-phosphate depends on its specific cellular location. In skeletal muscle cells, it is primarily utilized in glycolysis and ATP production. However, in hepatocytes, glucose-6-phosphate is converted into glucose, which is then released into systemic circulation.
Interestingly, glycogen degradation and synthesis processes are not typically occurring simultaneously under normal physiological conditions. Cells employ unique regulatory mechanisms to manage these metabolic processes in a reciprocal manner.
While the role of insulin in promoting glycogen synthesis in a fed state has been discussed, glucagon plays a crucial role in facilitating glycogen degradation. Upon binding to its receptor, glucagon initiates a downstream cascade, leading to the production of cyclic AMP (a second messenger). This molecule then binds to regulatory sites on Protein Kinase A (PKA), effectively inhibiting glycogen synthase (involved in glycogenesis) while simultaneously activating glycogen phosphorylase (involved in glycogenolysis).
Mechanisms of Action of metformin
Mitochondria (Within Hepatocyte) – Metformin acts to inhibit complex I of the Electron Transport Chain, thereby reducing ATP production. This creates an increase in ADP:ATP and AMP:ATP ratios. This deficiency in cellular ATP is recognized by AMPK, the “cellular energy sensor”. Consequently, AMPK works to promote catabolic processes that generate ATP while concurrently inhibiting anabolic processes that consume ATP. The overall glycemic outcome is a reduction in hepatic glucose output.
Cytoplasm (Within Hepatocyte) – Metformin acts to inhibit the enzyme fructose 1,6 bisphosphatase, which plays a critical role in gluconeogenesis. This inhibitory action is initiated by the AMP:ATP ratio induced by metformin and results in an acute inhibition of gluconeogenesis. This pathway operates independently of AMPK. – The overall glycemic outcome is a reduction in hepatic glucose output.
Gastrointestinal Tract (Within Colonic Enterocyte) – Metformin stimulates an increase in anaerobic glucose metabolism within enterocytes.
Skeletal Muscle – Metformin facilitates the uptake of glucose by glucose transporters, specifically GLUT-4.
Practice Guide for Metformin
Metformin is a synthetic analog of guanidine, a natural compound isolated from the plant Galega officinalis, commonly known as the French lilac plant. This plant had medicinal applications in medieval Europe where it was used to treat a condition marked by “sweet tasting urine” – a condition we now recognize as diabetes mellitus.
The typical starting dosage for Metformin is set at 500mg per day, which can be gradually increased by increments of 500mg each week until a common maintenance dose of 1000mg twice daily is reached.
One of the known side effects of Metformin includes gastrointestinal discomfort and diarrhea, which can be mitigated by incrementally increasing the dosage at the beginning of the treatment regimen.
Given the potential for Metformin-associated lactic acidosis (MALA), it is crucial to adjust the dosage of Metformin for patients diagnosed with chronic kidney disease.
60 mL per minute per body surface area – No adjustment necessary, monitor renal function on an annual basis
45 to <60 mL per minute per body surface area – No adjustment necessary, monitor renal function every three to six months
30 to 45 mL per minute per body surface area – Reduce to 500mg per day. Do not start metformin treatment in patients without prior exposure.
< 30 mL per minute per body surface area – Usage of Metformin is contraindicated
References
- LaMoia TE, Shulman GI (2021) Cellular and Molecular Mechanisms of Metformin Action. Endocr Rev 42:77–96
- Gunton JE, Delhanty PJD, Takahashi S-I, Baxter RC (2003) Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab 88:1323–1332
- Hardie DG, Ross FA, Hawley SA (2012) AMPK – a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262
- Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia 60:1577–1585
- Yendapally R, Sikazwe D, Kim SS, Ramsinghani S, Fraser-Spears R, Witte AP, La-Viola B (2020) A review of phenformin, metformin, and imeglimin. Drug Dev Res 81:390–401
- Zhou J, Massey S, Story D, Li L (2018) Metformin: An Old Drug with New Applications. Int J Mol Sci 19:2863
- Davidson MB, Peters AL (1997) An overview of metformin in the treatment of type 2 diabetes mellitus. Am J Med 102:99–110
- Blough B, Moreland A, Mora A (2015) Metformin-induced lactic acidosis with emphasis on the anion gap. Proc Bayl Univ Med Cent 28:31–33
- Hur KY, Kim MK, Ko SH, Han M, Lee DW, Kwon HS, Committee of Clinical Practice Guidelines, Korean Diabetes Association, Committee of the Cooperative Studies, Korean Society of Nephrology (2020) Metformin Treatment for Patients with Diabetes and Chronic Kidney Disease: A Korean Diabetes Association and Korean Society of Nephrology Consensus Statement. Diabetes Metab J 44:3–10
- DeFronzo RA, Goodman AM (1995) Efficacy of Metformin in Patients with Non-Insulin-Dependent Diabetes Mellitus. N Engl J Med 333:541–549