
Calcium is the most abundant mineral in the human body, with about 99% found in bone. It is essential in several biochemical processes, including hormone secretion, bone mineralization, maintenance of cell integrity, coagulation of blood, neuromuscular contraction, and signal transduction. Several hormones with regulatory effects on the parathyroid glands, intestine, bone, and kidneys ensure the maintenance of serum calcium within a narrow physiologic range. The element phosphorus plays several functions in all biological systems.
It forms a critical component of nucleic acids, the building blocks of our DNA, and is essential in metabolism due to its central role in the activation and inactivation of enzymes. Calcium and phosphorus metabolism shares essential regulatory hormones, for example, PTH, fibroblast growth factor, and calcitriol. Thus, perturbations in serum levels of either of these minerals can affect the serum concentration of the other.
Discovery of the parathyroid gland
Richard Owen first described the parathyroids in a paper published in 1862. Owen, a professor of comparative anatomy in London, described the parathyroid gland as “a small yellow body attached to the thyroid” during an autopsy of a rhinoceros. However, the discovery of the parathyroid glands has been credited to Ivar Sandström, who published his discovery in 1880 in a paper titled “On a New Gland in Man and Several Animals.” Due to the late anatomical description of these glands, they have often been referred to as “the last anatomic discovery.”
Physiologic states of calcium and phosphate
Approximately 1% of total body calcium is outside the skeletal system. The amount of calcium present in soft tissues and extracellular fluid compartments is accounted for by this relatively insignificant proportion of total body calcium. The extracellular fluid compartment comprises unbound (free) calcium and bound (complexed to albumin and organic molecules) calcium in almost equal proportions.
About 50% of extracellular calcium is “ionized” and biologically active, 40% remains albumin-bound, and 10% is bound to circulating anions. Interestingly, the common term for free calcium, i.e., ionized calcium, is a misnomer since all the calcium in the human body is ionized. Indeed, “ionized calcium” in plasma or serum exists in a hydrated state and not as a freely circulating divalent cation.
Over 80% of total body phosphate is bound to hydroxyapatite crystals in the skeleton. The rest of it is present in the viscera and skeletal muscle. Phosphate ions in the extracellular fluid compartment account for less than a 10th of the percentage of total body phosphate.
Regulation of ionized calcium based on acid-base balance
Changes in pH impact the availability of albumin binding sites, which invariably determines the fraction of unbound calcium. Metabolic perturbations in acid-base balance correlate with the levels of ionized calcium. Metabolic alkalosis causes an increase in circulating bicarbonate which binds ionized calcium.
In addition, the binding of circulating albumin with calcium increases in the setting of alkalosis. Both reasons account for the low level of ionized or free calcium in the setting of alkalosis. The converse occurs in the setting of metabolic acidosis. There is a relatively lower level of circulating bicarbonate; this increases the fraction of ionized or free calcium since less calcium is bound by bicarbonate. Decreased binding of ionized calcium to albumin occurs during metabolic acidosis as well; this is another reason for hypercalcemia in the setting of acidemia.
The role of the calcium-sensing receptor activation in mediating serum calcium
Calcium Sensing receptor (CaSR) in the parathyroid gland: CaSR present on the cellular membrane of the chief cells of the parathyroid gland senses the level of extracellular calcium and regulates calcium concentration by influencing PTH synthesis.
Increasing amounts of extracellular ionized calcium activate the CaSR, which sets off a cascade of intracellular reactions (Phospholipase C and adenylate cyclase mediated processes), which increases cytosolic calcium concentration. Calcium Response Elements (CRE) present on the PreProPTH gene detect the high intracellular calcium and downregulate the transcription and translation of PTH. Therefore, the net effect of CaSR activation in the parathyroid gland is a reduction in PTH synthesis.
Calcium Sensing receptor (CaSR) in the kidney: The CaSR on the basolateral surface of the thick ascending limb of Henle’s loop (TALH) is activated in the setting of hypercalcemia. Through various intracellular processes, there is a downregulation of potassium channels and the sodium-potassium adenosine triphosphatase (Na-K-ATPase). The necessary luminal positive electrical gradient required for the absorption of divalent cations such as magnesium and calcium is therefore impaired. This results in hypercalciuria and a reduction in serum calcium. The net effect of CaSR activation in the TALH is, therefore, a reduction in serum calcium.
Familial hypocalciuric hypercalcemia is an autosomal dominant disorder characterized by a loss of function mutation of the CaSR gene. The CaSR is, therefore, unable to sense the levels of ionized calcium in the extracellular fluid. At the level of the parathyroid gland, this results in PTH synthesis even in the setting of hypercalcemia. In the TALH, there is increased calcium conservation, which leads to mild hypercalcemia.
Most patients with this condition are asymptomatic but may occasionally develop acute pancreatitis or cholelithiasis. An important endocrinology dictum requires all patients undergoing evaluation for primary hyperparathyroidism to have an assessment of 24-hour urinary calcium excretion. This will prevent inadvertent parathyroid gland exploration in patients with hypocalciuria in the setting of PTH-mediated hypercalcemia.
Autosomal dominant hypocalcemia with hypercalciuria (ADHH) is characterized by a gain of function mutation of the CaSR. As previously mentioned, CaSR activation in the chief cells of the parathyroid gland reduces PTH synthesis and promotes hypercalciuria in the TALH. The biochemical phenotype is, therefore, similar to that of idiopathic hypoparathyroidism, i.e., hypocalcemia and hyperphosphatemia; however, with a low but detectable PTH. Patients with ADHH develop significant suppression of PTH after calcium supplementation and are generally at an increased risk for nephrocalcinosis.
The effect of calcium-sensing receptor mutations on PTH secretion and renal calcium conservation
| Pathophysiology | PTH | Serum calcium | |
| FHH | Loss-of-function mutation of the CaSR | Increased PTH synthesis and secretion | Hypercalcemia due to increased renal calcium reabsorption |
| ADHH | Gain-of-function mutation of the CaSR. | Decreased PTH synthesis and secretion | Hypocalcemia due to reduced renal calcium reabsorption |
ADHH Autosomal dominant hypocalcemia with hypercalciuria, FHH Familial hypocalciuric hypercalcemia, CaSR Calcium sensing receptor, PTH Parathyroid hormone
ADHH Autosomal dominant hypocalcemia with hypercalciuria, FHH Familial hypocalciuric hypercalcemia, CaSR Calcium sensing receptor, PTH Parathyroid hormone
Intestinal Handling of Calcium and Phosphate
Calcium is acquired through ingestion of dietary products, including dairy (milk, cheese, yogurt), vegetables, legumes, and animals, among other foods. Therefore, the intestine serves as the only portal of calcium entry into the human body. Up to 1 g of calcium is ingested daily, of which approximately 25% (200mg) is fractionally absorbed. Calcium absorption in the small intestine occurs via active carrier-mediated calcium transport (transcellular) and concentration-dependent passive diffusion processes (paracellular).
Transcellular transport of calcium from the luminal to the serosal side of the intestine is more prevalent in the duodenum and jejunum. Paracellular processes responsible for moving calcium from the mucosa to serosa tend to occur in the jejunum and ileum and depend on an electrochemical gradient. Intestinal absorption of calcium is regulated by 1,25(OH)2D3 (calcitriol). Calcitriol causes changes in the structure and function of enterocytes after binding to its intracellular vitamin D receptor (VDR).
The net effect is an increase in intestinal calcium absorption. Recent evidence suggests that both the paracellular and transcellular pathways of calcium absorption are mediated by multiple hormones, including thyroid hormone, growth hormone, estrogen, and prolactin, although the exact mechanisms have not been fully elucidated.
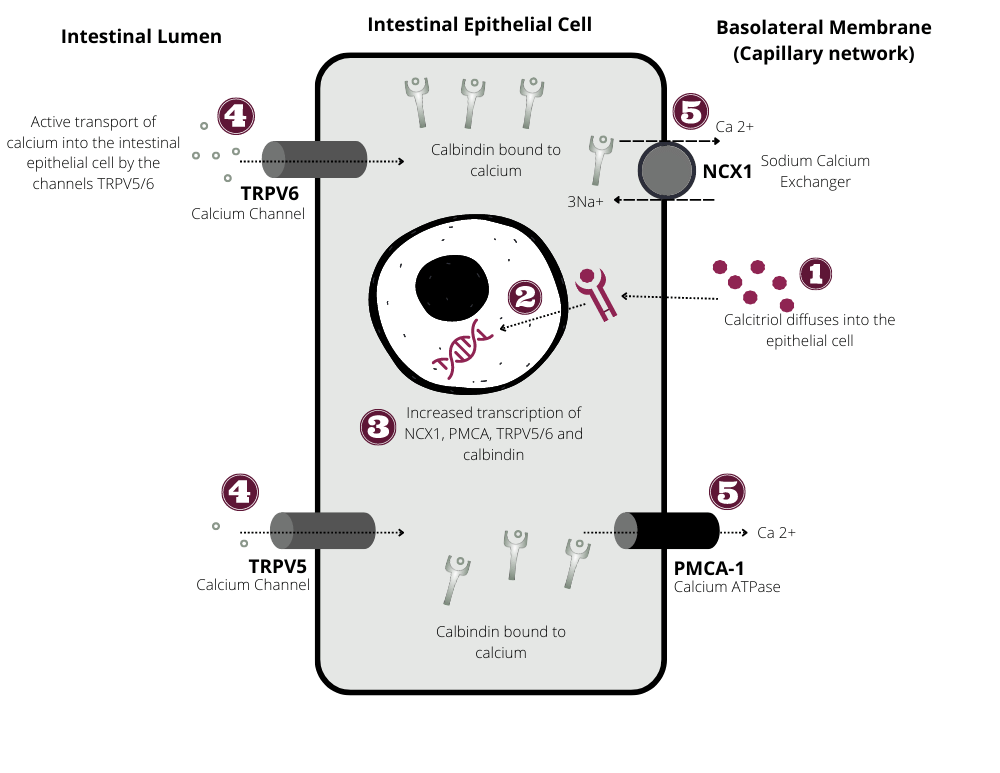
Schematic representation of intestinal absorption of calcium and phosphate
Calcitriol-mediated intestinal calcium absorption: Calcitriol binds to its intracellular vitamin D receptor (VDR). The calcitriol–VDR complex then binds to hormone response elements (HREs) on DNA and induces transcription of multiple calcium channels (TRPV6, TRPV5), and electrolyte exchangers (NCX1), calcium ATPase (PMCA1), and calcium-binding proteins, calbindin (CB). Insertion of calcium channels on the apical membrane of the enterocyte allows the influx of calcium into the enterocyte. CB carries calcium from the apical to the basolateral side of the enterocyte. Calcium-ATPase, an ATP-dependent transporter, is involved in pumping calcium out of the cytosol into the capillary network on the basolateral side of the intestinal epithelial cell.
The average daily dietary intake of phosphorus has been reported to range from 800 to 1,200 mg. Up to 70% of this amount is absorbed in the jejunum and also involves both transcellular and paracellular pathways. There are sodium-dependent phosphate (NaPi) cotransporters on the luminal membrane of
the enterocyte involved in the transcellular transfer of inorganic phosphate. Similar to calcitriol-mediated calcium absorption, calcitriol binds to its nuclear vitamin D receptor and results in increased transcription of NaPi cotransporters. Similarly, PTH regulates the transcription and insertion of Na-
Pi transporters on the luminal membrane of the enterocyte. Unlike transcellular phosphate transport, the paracellular route is not under hormonal control. Paracellular transport is unsaturable and dependent on dietary phosphate load.
Renal Handling of Calcium and Phosphate
There are three principal sites of renal calcium handling: the proximal tubule, the thick ascending limb of the loop of Henle (TALH), and the distal convoluted tubule (DCT). Approximately 60 to 70% of calcium absorption occurs through mainly passive diffusion processes at the proximal tubule of the nephron. An additional 25 and 10% of renal calcium conservation occurs at the TALH and DCT, respectively. A passive, nonhormone-dependent process accounts for up to 80% of calcium reabsorption in the proximal tubule.
Active transcellular transport of calcium is responsible for an additional 10 to 15% of calcium reabsorption at the proximal tubule and is regulated by PTH. Calcium-sensing receptors expressed in the basolateral membrane of the TALH conserve filtered urinary calcium load through a PTH-independent process.
Calcium reabsorption at the distal renal tubule is dependent on an active transport system, which transfers calcium from the luminal to the basolateral side of the tubular cell. Active calcium transport at the DCT occurs against an electrochemical gradient and is tightly regulated by calcitriol.
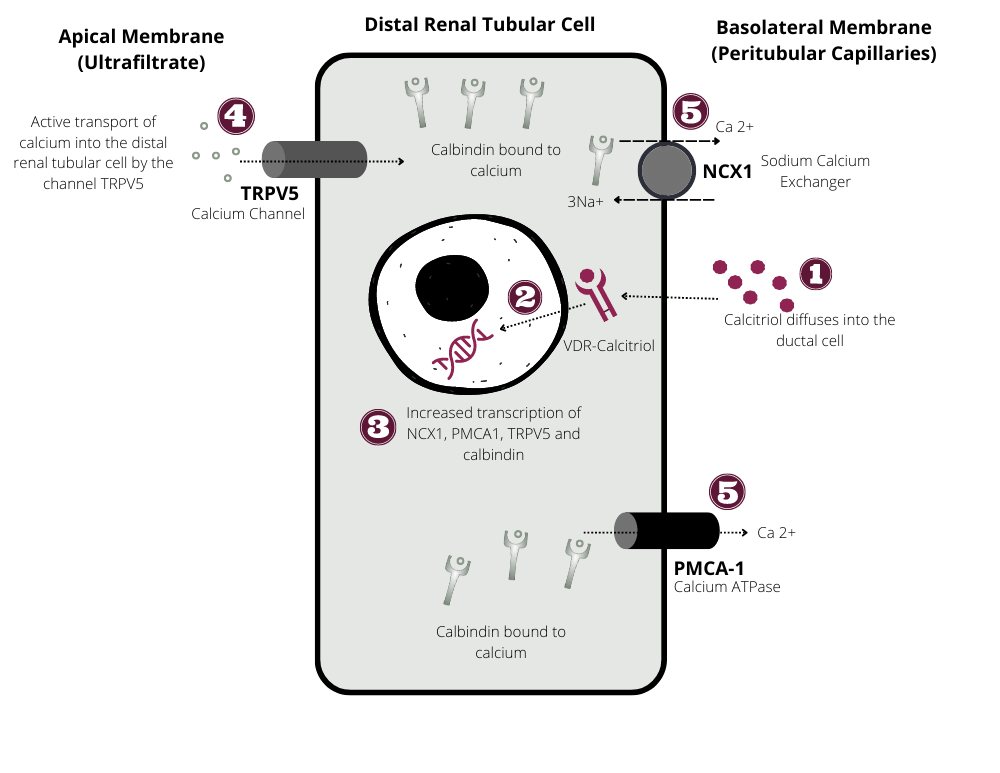
Schematic representation of renal absorption of calcium and phosphate
Calcitriol-mediated calcium absorption at the distal renal tubule: Calcitriol diffuses through the basolateral membrane of the distal renal tubular cell to bind its cytosolic vitamin D receptor (VDR). The calcitriol–VDR complex binds to hormone response elements (HREs) on DNA to initiate transcription of calcium channels (TRPV5), calcium exchangers (NCX1), and calcium ATPase (PMCA1). Calcium channels (TRPV5) present on the apical membrane are involved in the direct translocation of calcium from the renal tubule into the cytosol of the distal renal tubular cell. This step is then followed by cytosolic transfer of ionized calcium by calbindin (calcium-binding protein) to the basolateral surface of the distal tubular cell. The extrusion of calcium into the peritubular vasculature is facilitated by NCX1 and PMCA1.
A significant amount of renal phosphate absorption, approximately 85%, occurs at the proximal tubule and involves an active transport system. Other mechanisms underlying renal phosphate conservation are yet to be elucidated. The calcitriol–VDR complex, through its effects on gene transcription, results in an increased expression of sodium-dependent phosphate transporters in the luminal membrane of the proximal tubular cell. Phosphate is transported from the luminal membrane, through the cytosol of the tubular cell, to the basolateral capillary network.
Storage of calcium and phosphate.
Bone is composed of inorganic salts made up of calcium and phosphorus existing in hydroxyapatite Ca10(PO4)6(OH)2 crystals and an organic matrix consisting of mainly collagenous and noncollagenous proteins. Approximately 1 kg of calcium is stored up in the mineralized portion of our bones. This makes the human skeleton the largest repository of calcium. The total body phosphorus content is about 700 g, of which 85% is present in the skeletal system.
Hormonal control of calcium and phosphate regulation
Calcitriol
The synthesis of calcitriol, the metabolically active form of vitamin D, starts in the epidermis of the skin. Ultraviolet light from the sun converts 7-dehydrocholesterol in the skin into a prohormone called previtamin D. Previtamin D subsequently goes through a nonenzymatic isomerization step to be converted to vitamin D3. Vitamin D3 is then transferred from the skin to the liver, where it undergoes an intermediate conversion into 25-hydroxyvitamin D, before being released into the circulatory system.
A storage form of vitamin D, 25-hydroxyvitamin D, is bound to plasma proteins such as albumin and vitamin D-binding protein. The renal 1α-hydroxylase enzyme present in the proximal convoluted tubule converts 25-hydroxyvitamin D to 1,25-hydroxyvitamin D (calcitriol). Calcitriol, the metabolically active form of vitamin D, is involved in calcium and phosphate regulation at the level of the intestine, kidneys, and bone. Calcitriol also promotes the synthesis of FGF-23, a critical regulatory enzyme involved in negative feedback inhibition of calcitriol synthesis.
Parathyroid hormone
PTH has an inverse relationship with ionized (free) calcium. High levels of circulating calcium are sensed by G-protein coupled calcium-sensing receptors (CaSRs) present in the parathyroid gland. Calcium binding to the CaSR initiates a cascade of second messenger systems, which results in the regulation of the secretion of PTH from the parathyroid glands. PTH has multiple effects on the skeleton, kidney, and intestine.
PTH promotes calcium absorption in the TALH and DCT of the nephron. It inhibits phosphate absorption by impairing the insertion of sodium-phosphate cotransporters into the luminal membranes of the proximal and distal convoluted tubule.
Through its stimulatory effects on renal 1α-hydroxylase enzyme activity, PTH promotes the formation of calcitriol. As has been previously described, calcitriol is a regulatory hormone involved in intestinal calcium and phosphate conservation. PTH promotes bone resorption by binding to PTH receptors on osteoblasts, leading to their activation. Osteoclast activation promotes bone resorption and mobilization of calcium from skeletal stores.
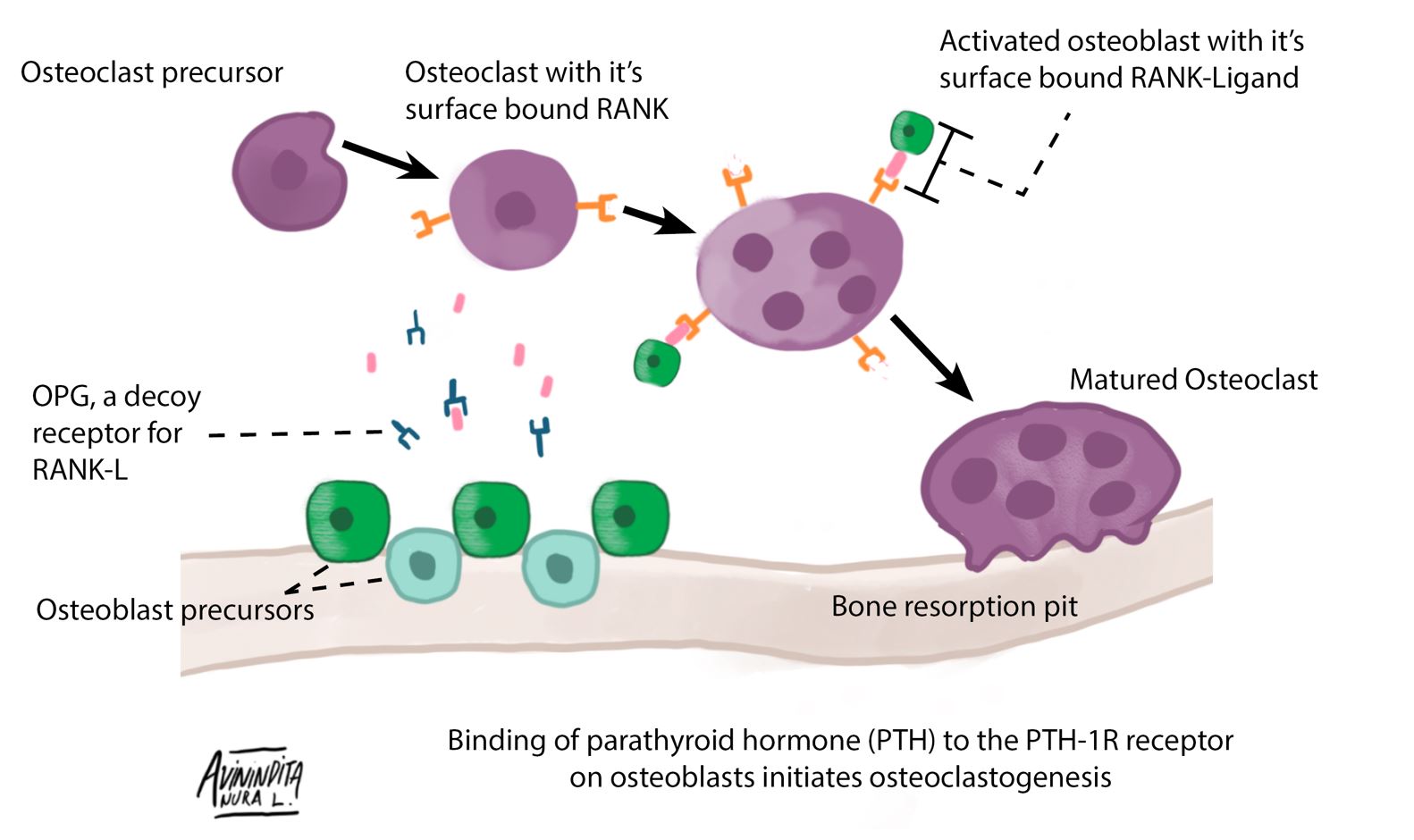
Osteoclastogenesis
Role of parathyroid hormone (PTH) in osteoclastogenesis: PTH binds to the PTH-1 R receptors present on osteoblasts, leading to their activation. Osteoblast surface-bound receptor activator of nuclear factor-kappaB ligand (RANKL) binds to receptor activator of nuclear factor-kappaB (RANK) present on osteoclast precursors. This RANK–RANKL interaction leads to activation and differentiation of the osteoclast precursor into a matured osteoclast. Osteoprotegerin, the decoy receptor for RANKL, is expressed by osteoclasts and provides negative feedback inhibition of the process of RANK–RANKL interaction.
Calcitonin
Calcitonin, a hormone produced by the C cells of the thyroid, is released in response to hypercalcemia. Calcitonin reduces calcium resorption in bone through its inhibitory effects on osteoclastogenesis. There is evidence that it decreases renal tubular absorption of calcium. Its protective effect on hypercalcemia is rather mild; it, therefore, has an insignificant contribution to calcium homeostasis.
The metabolic effects of calcitonin are poorly understood, and its deficiency is not associated with any adverse metabolic outcomes. Excess secretion of calcitonin, such as those that might occur in patients with medullary thyroid carcinoma, can result in diarrhea.
Fibroblast growth factor 23
The FGF-23–kidney–bone axis is a recently discovered physiologic pathway in calcium and phosphate metabolism. Calcitriol activates the release of FGF-23 from osteocytes and osteoblasts. FGF-23 suppresses the expression and subsequent insertion of sodium-phosphate cotransporters on the luminal side of the proximal convoluted tubule; this accounts for the phosphaturic effect of FGF-23. It also reduces the circulating levels of active vitamin D (calcitriol) through its inhibitory and stimulatory effects on 1α-hydroxylase and 24-hydroxylase enzymes, respectively. The net effect is impaired calcium and phosphate conservation.
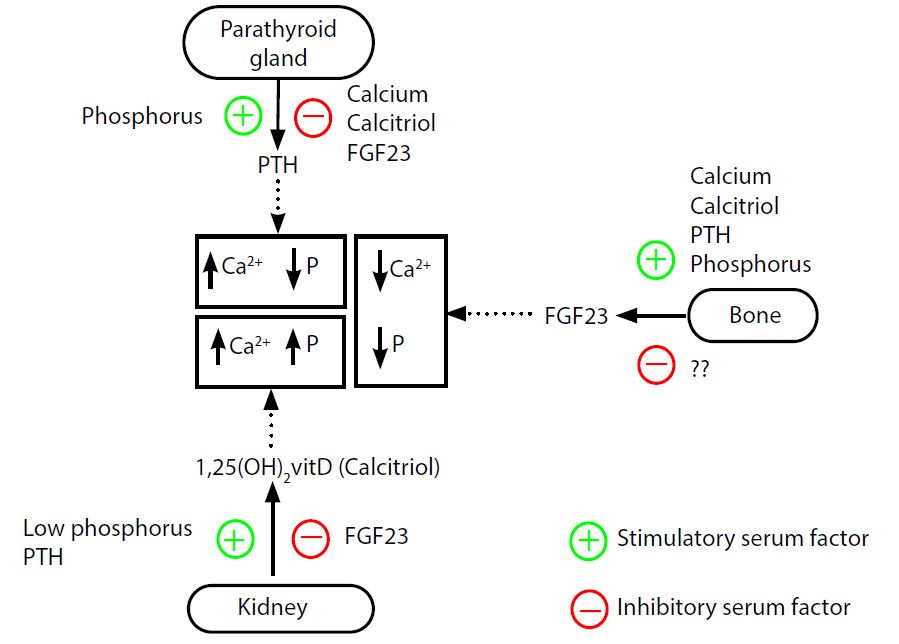
Role of parathyroid hormone, fibroblast growth factor 23 (FGF-23), and calcitriol in calcium and phosphate balance. This figure summarizes the roles of the various hormones involved in calcium and phosphate metabolism. The skin, liver, kidney, and bone serve as vital target tissues for these hormones.
Clinical Pearls
A 23-year-old Caucasian female with a 2-year history of amenorrhea and galactorrhea presented to the emergency room with a 2-day history of colicky abdominal pain suggestive of renal colic. A renal sonogram was significant for bilateral nephrolithiasis with an obstructing left ureteric stone requiring placement of a stent. Her serum calcium was elevated at 12.5 with an estimated glomerular filtration rate (GFR) of 23. Her hypercalcemia at presentation was attributed to acute kidney injury in the setting of obstructive uropathy. The patient’s basic metabolic panel repeated 48 hours after adequate intravenous hydration showed:
● Serum calcium of 11.2 (8.3–10.5 mg/dL)
● Serum creatinine 0.6, estimated GFR > 60
During her 2-week post-hospital discharge follow-up at the clinic, she was noted to have serum calcium of 12.1 with an estimated GFR > 60. A review of the patient’s chart revealed calcium levels between 10.7 and 11.3 in the preceding 2 years. The surgical team requested a serum PTH level, 25-hydroxyvitamin D, ionized calcium, and calcium to creatinine excretion ratio. PTH was normal at 57 (15–65), ionized calcium was elevated at 1.57 (1.1–1.35 mmol/L), and 25-hydroxyvitamin D was optimal at 35 (30–100). The calcium to creatinine excretion ratio was 0.05 (< 0.01 is suggestive of hypocalciuria).
What is the diagnosis of this patient?
Primary hyperparathyroidism. The patient has an inappropriately elevated serum PTH in the setting of hypercalcemia.
Rationale
Serum calcium has an inverse relationship with PTH levels. The CaSRs of the parathyroid gland sense hypercalcemia, and this ultimately results in suppression of PTH secretion in normal physiology. Clinicians often overlook the concept of “inappropriately normal” PTH due to the expected elevation of PTH in the setting of autonomous PTH secretion by a parathyroid adenoma (primary hyperparathyroidism). It is important to note that, in the context of hypercalcemia which is not PTHmediated, we should expect marked suppression of PTH with levels below the reference range or much closer to the lower limit of the reference range. In this patient with a PTH level close to the upper limit of normal despite being hypercalcemic, a normal PTH is inappropriate and represents a pathologic state. The patient’s clinical presentation is therefore highly suggestive of primary hyperparathyroidism.
Try this quiz on calcium and parathyroid metabolism
References
- Pu F, Chen N, Xue S. Calcium intake, calcium homeostasis and health. Food Sci HumWellness. 2016; 5(1):8–16
- Greco DS. Endocrine causes of calcium disorders. Top Companion Anim Med. 2012; 27(4):150–155
- Quinn SJ, Thomsen ARB, Pang JL, et al. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab. 2013; 304(3):E310–E320
- Bailey RL, Dodd KW, Goldman JA, et al. Estimation of total usual calcium and vitamin D intakes in the United States. J Nutr. 2010; 140(4):817–822
- Pansu D, Bellaton C, Bronner F. Effect of Ca intake on saturable and nonsaturable components of duodenal Ca transport. Am J Physiol. 1981; 240(1):G32–G37
- Kim BK, Lee J, Sun WY. Recurrent hyperparathyroidism due to proliferation of autotransplanted parathyroid tissue in a multiple endocrine neoplasia type 2A patient. Ann Surg Treat Res. 2016; 91(3):145–148
- Thakker RV, Newey PJ, Walls GV, et al. Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012; 97(9):2990–3011