Structure Of Insulin
Insulin is a 51 amino acid peptide hormone that consists of two chains (A and B) linked by a pair of disulfide bonds. The A (21 amino acids) and B (30 amino acids) chains of insulin are linked by an intervening sequence of amino acids known as the connecting peptide (c-peptide)[1]. The gene that encodes human insulin produces an mRNA transcript that is translated into a large 110 amino acid polypeptide sequence known as preproinsulin. Preproinsulin then undergoes further processing in the lumen of the rough endoplasmic reticulum to produce proinsulin. Proinsulin is then ferried into the Golgi apparatus, where it is subsequently cleaved into the native insulin peptide and c-peptide [2]. Along with these products of proinsulin processing, amylin and other intermediate peptides are packaged into secretory granules by the pancreatic beta cell[3].

Structure of Insulin
Regulation of blood glucose
Glucose homeostasis depends on the action of insulin and a host of other counterregulatory hormones. This fine-tuned system depends on the action of hormones, neural stimuli, cytokines, and other regulatory cytokines working together in various organs to control plasma glucose. In fact, the pancreatic beta cell is crucial in directing the orchestra of this complex homeostatic system[4].
During the fasting state, the relative decrease in insulin levels results in the oxidation of fatty acids in adipose tissue, making fatty acids a primary source of energy during. For example, the liver uses fatty acids for gluconeogenesis during a prolonged fast. On the contrary, the brain has obligatory glucose requirements, which makes it necessary to have alternative sources of energy supply during a fast. The liver provides the needed glucose for the central nervous system by serving as a valuable source of glucose during a prolonged fast. Glucagon is released by alpha cells of the pancreas during fasting and promotes hepatic gluconeogenesis and glycogenolysis, thus maintaining plasma glucose concentration within a physiological range.
In contrast, during the postprandial state, glucose sensing by the pancreatic beta cell results in insulin release. Insulin then exerts its metabolic action through various processes, including the inhibition of hepatic glucose output (reduced glycogenolysis and gluconeogenesis), the promotion of glucose uptake by peripheral tissues (muscle and adipose tissue), and a reduction in lipolysis (adipose tissue)[5]. Next, we will explore the effects of insulin on various tissues, including skeletal muscle, adipose tissue, and liver.
Phases of insulin release after a meal
Insulin acts to lower high blood sugar levels by promoting the uptake of glucose into cells and inhibiting the liver’s production of glucose. Insulin release is a complex process that occurs in a biphasic manner (two distinct phases), known as the first and second phases of insulin release[6].
First Phase of Insulin Release: The first phase of insulin release occurs within minutes of a meal and is a rapid, short-lived response (lasting approximately 10 minutes) to an increase in arterial glucose concentration[7]. The rapid release of pre-formed secretory granules of insulin accounts for this phase. This initial surge of insulin is thought to be triggered by the stimulation of the beta cells in the pancreas by glucose and other nutrients in the meal. The first phase of insulin release helps to quickly lower blood glucose levels by promoting the uptake of glucose into cells and inhibiting hepatic glucose production[8].
Second Phase of Insulin Release: The second phase of insulin release occurs several minutes to several hours after the first phase and is a slower, sustained response to elevated blood glucose levels[3]. This phase of insulin release is thought to be regulated by the release of other hormones, such as incretins, that are produced in response to a meal. The second phase of insulin release helps to maintain stable blood glucose levels for several hours after a meal by promoting the uptake of glucose into cells and inhibiting the liver’s production of glucose[3,9].
Various circulating factors, including glucose, incretins, amino acids, ketone bodies, lactic acid, and fatty acids, facilitate the release of insulin by pancreatic beta cells. Furthermore, glucose-induced insulin release plays a predominant role in determining the concentration of extracellular insulin in both the fasting and postprandial state. Glucose enters pancreatic beta cells through the active glucose transporter 2 (GLUT-2) transporter. Glucokinase then catalyzes the metabolism of glucose into glucose-6-phosphate, which then results in the generation of intracellular ATP through the process of glycolysis. NADH and FADH2 produced from glycolysis are further incorporated into the mitochondrial electron transport chain to augment intracellular ATP levels[10]. Consequently, the high ATP to ADP ratio in the pancreatic beta cell inhibits the ATP-sensitive potassium channels in the plasma membrane[11]. This results in the depolarization of the plasma membrane, the opening of voltage-gated calcium channels, and the influx of calcium via the L-type voltage-gated calcium channels[12,13]. A rise in intracellular calcium then facilitates the fusion of insulin secretory granules with the pancreatic plasma membrane. Finally, insulin, c peptide, proinsulin, and other secretory products are released through exocytosis[3].

Schematic representation of insulin release from the pancreatic beta-cell.
Glucose transporter 2 channels ferry glucose into the pancreatic beta-cell. This is followed by the phosphorylation of glucose into glucose-6-phosphate by glucokinase. ATP generated as a consequence of glycolysis blocks potassium channels on pancreatic cell membranes, consequently depolarizing the cell membrane. Depolarization of the pancreatic beta-cell membrane activates pancreatic L-type voltage-gated calcium channels, which funnels calcium into the pancreatic beta-cell. Increased intracellular calcium leads to the liberation of endocytic stores of insulin into systemic circulation[14]. Fatty acids are an alternative insulin secretagogue. Fatty acids undergo beta oxidation and oxidative phosphorylation in pancreatic beta cells. ATP produced as a consequence then inhibits the ATP-dependent or sensitive potassium channels, which results in the eventual release of insulin via exocytosis (previously described as glucose-mediated insulin release)[15]. Also, fatty acids in the gut stimulate the release of incretins. Incretins then augment the release of insulin by pancreatic beta cells [9]
The effects of insulin on various tissues
Skeletal muscle relies on glucose and free fatty acids as energy sources in the postprandial and fasting states, respectively.
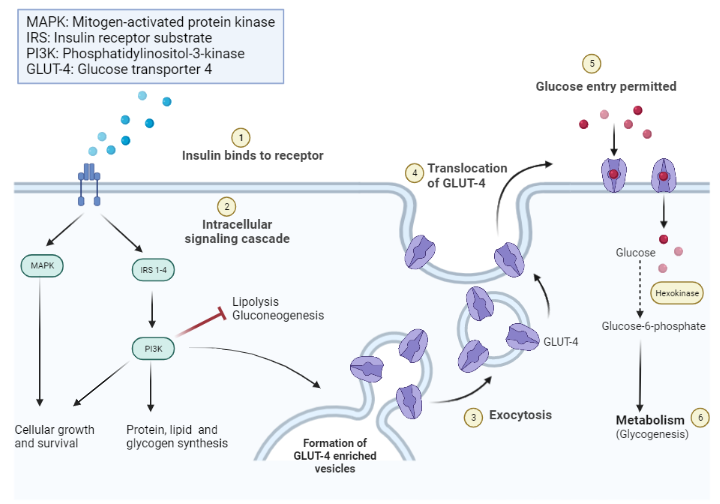
Skeletal muscle serves as the primary site of glucose uptake after ingestion of a meal (postprandial period), with insulin being the primary hormone responsible for this function. An increase in serum glucose after meal intake is sensed by pancreatic beta cells, which subsequently release insulin. Insulin then binds to the insulin receptor and initiates a signaling cascade that results in the ferrying of glucose transporter 4 (also known as GLUT 4) from the sarcoplasm to the plasma membrane of skeletal muscle [16,17] through a process of exocytosis[17]. GLUT-4 then mediates glucose uptake by the skeletal muscle.
When glucose enters the myocyte of skeletal muscles, it is phosphorylated by the hexokinase enzyme to glucose-6-phosphate, which can be channeled into either glycogen synthesis (and storage) or used in the glycolytic pathway. A substantial amount of glucose entering the glycolytic pathway is oxidized to produce energy, with only 10% being channeled into lactate production[18].
On the other hand, during the fasting state, relatively low insulin levels impair the usual anti-lipolytic action of insulin in white adipose tissue. Consequently, increased lipolysis in white adipose tissue results in the production of fatty acids. These free fatty acids become the primary source of fuel for skeletal muscle[19].
During fasting, the liver serves as the main source of endogenous glucose production. This allows cells that can only utilize glucose as a primary source of energy, such as neurons, red blood cells, and renal medulla cells, to function optimally. The liver achieves this goal by increasing glycogenolysis, gluconeogenesis, and glycogen synthesis in the postabsorptive period. This complex system requires the interaction of various hormones (insulin, glucagon, catecholamines, glucocorticoids), substrates (glucose, glycerol), and allosteric factors (acetyl CoA, glucose, and glucose-6-phosphate)[20].
In the postprandial period, insulin mediates various processes that suppress glucose production by the liver.
Through its antilipolytic action, insulin suppresses the breakdown of triglycerides into free fatty acids in white adipose tissue. Therefore, free fatty acid, a substrate for liver gluconeogenesis, is therefore reduced, resulting in the suppression of hepatic glucose production[20,21].
Glucagon stimulates hepatic glycogenolysis (increased hepatic glucose production). The presence of high levels of circulating insulin after a meal inhibits the release of glucagon by alpha cells of the pancreas which consequently reduces hepatic glucose output [21].
Insulin receptor
The insulin receptor (IR) has an extracellular portion composed of two alpha subunits and a transmembrane section comprising two beta subunits. In normal physiology, alpha subunits inhibit the intrinsic tyrosine kinase activity of beta subunits[22].
When insulin binds to the alpha subunit of the IR, it suppresses the ability of the alpha subunit to inhibit the beta subunit. Consequently, insulin binding to IR promotes the phosphorylation of intracellular proteins required for various downstream signaling cascades in different tissues[23].

Schematic diagram of the intracellular effects of insulin receptor activation.
Insulin-to-IR binding promotes intracellular effects, including increased glycogen, protein, and lipid synthesis. Lipolysis and gluconeogenesis are, however, inhibited. Insulin further promotes the recruitment of glucose transporter channels on peripheral tissues (e.g., skeletal muscle).
Diverse Metabolic Functions of Insulin
Insulin significantly influences a myriad of metabolic processes in body tissues. Among its many functions, a pivotal one is the regulation of blood glucose levels. It accomplishes this by facilitating the storage or utilization of glucose, thereby decreasing its concentration in the blood. Insulin facilitates glucose transport into specific tissues such as skeletal muscle, cardiac muscle and adipose tissue.
Transport of Glucose into Muscles and Adipose Tissues
In numerous tissues, glucose enters cells via insulin-independent GLUT proteins situated in cell membranes. Particularly in skeletal and cardiac muscle fibres and adipocytes, glucose initially enters cells to a limited extent via GLUT1 transporters. However, this transport pathway alone is inadequate for fulfilling the glucose needs beyond basal activity. As a result, the insulin-dependent GLUT4 transporters provide additional glucose, for instance, to actively contracting muscles. Furthermore, insulin actively stimulates the recycling of GLUT4 transporters from the intracellular cytoplasm to the plasma membranes and induces GLUT4 synthesis. Thus, insulin serves as a crucial regulator of this vital glucose entry pathway.
Stimulation of Glycolysis
Glycolysis involves a sequence of reactions that ultimately changes glucose into two pyruvate molecules, two ATP, two NADH, and two water molecules[24].

Comparison of glycolysis and gluconeogenesis.
Glucose is converted to glucose-6-phosphate via the enzyme hexokinase. The formation of glucose-6-phosphate destabilizes the glucose molecule and traps it inside the cell. Phosphoglucose isomerase then facilitates an isomerization step that converts glucose-6-phosphate to fructose-6-phosphate. Phosphofructokinase then promotes the phosphorylation of fructose-6-phosphate into fructose-1,6-bisphosphate. Sequential enzymatic steps ultimately result in the formation of pyruvate[24]. Insulin also initiates glycolysis by stimulating the transcription of three key enzymes that control essential steps in glucose metabolism. These enzymes are glucokinase, phosphofructokinase, and pyruvate kinase. It’s important to note that insulin stimulates all cells, not just muscle and adipose tissue cells, to enhance this process.
Promotion of Glycogenesis
Glycogenesis is the process of the formation of glycogen molecules (a storage form of glucose). After ingesting a meal rich in carbohydrates, plasma glucose levels rise. Pancreatic beta cells detect this increase in plasma glucose and, in turn, release insulin. Insulin then travels through the bloodstream to hepatocytes, binding to its corresponding insulin receptor and initiating a signal transduction pathway that activates various protein kinases in the cytoplasm[25].
These protein kinases inactivate glycogen synthase kinase (an enzyme that is responsible for the inactivation of glycogen synthase). Consequently, glycogen formation is promoted due to the loss of inhibition of the key regulatory enzyme, glycogen synthase[26].

Insulin-mediated glycogen synthesis
Upon insulin binding to its receptor, a signaling cascade is initiated, involving the activation of insulin receptor substrate (IRS) and phosphatidylinositol 3-kinase (PI3K). This leads to the activation of Akt (protein kinase B), which inhibits glycogen synthase kinase-3 (GSK-3). The inhibition of GSK-3 promotes the conversion of inactive glycogen synthase b to its active form, glycogen synthase a, facilitating glycogen synthesis and storage[27]. Glycogen synthase ultimately catalyzes the elongation of glycogen by sequentially attaching glucose monomers via alpha 1,4 glycosidic bonds[28]. When glucose enters a cell, it is quickly phosphorylated to glucose-6-phosphate and is then either immediately used by the glycolytic pathway or stored as glycogen. Insulin promotes this conversion of glucose-6-phosphate to the complex branched polysaccharide glycogen via the process of glycogenesis. Insulin stimulates glycogenesis in all cells, including hepatocytes, which serve as a significant glycogen storage site and hence a ready source of glucose in the body.
Inhibition of Glycogenolysis
Glycogenolysis occurs in the liver, the glucose is released into the general circulation, making it available for use by other cells in the body. It is noteworthy that insulin inhibits the enzymes involved in glycogenolysis, further illustrating its complex role in glucose metabolism.
Inhibition of Gluconeogenesis by Insulin
The normal human body requires, on average, about 160 grams of glucose per day[29], of which 75% is utilized primarily by the central nervous system for energy production[30]. There are, however, very limited stores of readily accessible glucose in humans. Indeed, only 20 grams of glucose is present in body fluids, with almost 190 grams present in a stored form known as glycogen[31].
Depending on the level of energy expenditure by an individual, additional sources of glucose may be required. Furthermore, survival beyond 24 hours without extra dietary glucose intake would be impossible since most of these reserves would be depleted[32]. This makes gluconeogenesis a critical process for producing valuable glucose molecules needed for energy production. Gluconeogenesis generates glucose endogenously from mainly non-glucose precursors such as pyruvate, lactate, glycerol, and amino acids. It occurs mainly in hepatocytes and renal tissue, with these sites accounting for 75% and 25% of glucose produced through gluconeogenesis, respectively[33,34].
Sources of glucose precursors required for gluconeogenesis.
Glucose precursor | Source |
Lactate | It is produced by anaerobic respiration in skeletal muscle. Lactate is then transformed into pyruvate by lactate dehydrogenase in the liver. |
Glycerol | It is produced by the breakdown of triglycerides in adipose tissues. Glycerol is then converted into dihydroxyacetone phosphate (DHAP) in readiness for gluconeogenesis. |
Amino acids | Hydrolysis of protein in food or skeletal muscle generates amino acids, which are converted either into pyruvate or DHAP. |
Based on reference [35]
The first two steps of gluconeogenesis aim to convert pyruvate into phosphoenolpyruvate (PEP), with oxaloacetate being an intermediate product. These steps required ATP and carbon dioxide[36]. Once phosphoenolpyruvate is formed, a series of sequential enzymatic (steps 3-7) results in the formation of fructose 1,6 bisphosphate. Steps 3-7 represent a complete reversal of exact pathways seen in glycolysis.
Fructose 1,6 bisphosphate is hydrolyzed into fructose 6-phosphate under the influence of the allosteric enzyme, fructose 1,6 bisphosphate. Subsequently, fructose 6-phosphate then undergoes an isomerization step which produces glucose-6-phosphate mediated by phosphoglucose isomerase. The fate of glucose-6-phosphate varies significantly by the tissue in which it is formed. Unlike glucose, glucose-6-phosphate cannot permeate cellular membranes, thus making it an easily accessible substrate for further metabolic processes.
In skeletal muscle, for example, glucose-6-phosphate can be turned into either glycogen (a storage form of glucose) or pyruvate to be utilized in energy production (glycolysis)[33,37].
Conversely, in hepatocytes and renal cells, glucose-6-phosphate is converted to glucose in the presence of the enzyme glucose-6-phosphatase. Since cellular membranes contain glucose transporters, glucose can readily leave these primary sites of gluconeogenesis in order to be transported to other cells in the human body[38].
Insulin acts as an inhibitor of gluconeogenesis, the synthesis of glucose from non-carbohydrate precursors such as specific amino acids and glycerol. This metabolic process mainly occurs in the liver, and insulin’s role in its inhibition helps prevent persistent hyperglycemia.
Insulin’s Indirect Influence on Glucose Production through Glucagon Inhibition
Another indirect mechanism by which insulin impacts glucose production is through the inhibition of glucagon release, largely attributed to a paracrine effect. When insulin levels are deficient or entirely absent, as seen in diabetes mellitus, glucose levels typically rise, and paradoxically, glucagon levels are often higher than expected. One possible explanation is the lack of the usual inhibitory influence exerted by insulin on the α-cells that produce glucagon.
Insulin’s Role in Protein Metabolism
Insulin can be characterized as an anabolic hormone due to its promotion of protein synthesis. This includes its influence on both amino acid uptake and protein synthesis. There exist various specific transporters that aid the passage of amino acids across cell membranes, some of which are stimulated by insulin. For example, the cationic amino acid transporter (CAT1) for arginine and the transporter for alanine are both directly stimulated by insulin. These substrates are then utilized in protein synthesis.
Beyond its likely stimulation of protein synthesis through its influence on amino acid transporters, insulin also induces new protein synthesis via an indirect genomic effect. Hence, insulin contributes significantly to normal body growth and development.
Influence of Insulin on Lipid Metabolism
Lipogenesis, the process through which glucose is converted to lipid in adipose tissue, offers an additional metabolic pathway for glucose storage. As glucose enters the glycolysis pathway, its end product, pyruvate, can be converted to acetyl co-enzyme A (acetyl CoA) in the cytoplasm. This is then converted to fatty acids by a complex of enzymes known collectively as fatty acid synthase. Three free fatty acids are esterified with one molecule of glycerol to form a triglyceride, which is subsequently transported and stored as fat. Insulin stimulates the synthesis of fatty acids and the formation of triglycerides, which are deposited in various tissues, particularly in adipose tissue. Simultaneously, insulin inhibits lipolysis, the breakdown of triglycerides back to their constituent fatty acids and glycerol, underscoring its role as a glucose storage hormone
Insulin’s Influence on Ketogenesis
Acetyl co-A can be converted to acetoacetyl co-A in adipocytes, especially when lipolysis is promoted in the absence of insulin. Acetoacetyl co-A is then transformed into hydroxyl-β-methylglutaryl co-A, which is subsequently converted into acetoacetic acid. The products of this process, acetoacetic acid and its two metabolites – acetone and β-hydroxybutyric acid, which are collectively known as the ketone bodies.
Normally, insulin, in the presence of glucose, suppresses the formation of these ketone bodies. However, in the minimal presence or absence of insulin, such as in type 1 diabetes mellitus, the concentration of ketone bodies in the blood increases.These acidic molecules can impair cellular function as they promote significant acidemia. The excretion of acetone, detectable by its characteristic nail varnish smell, in a patient with Kussmaul respiration, can serve as an important clinical clue of diabetic ketoacidosis.
Central Actions of Insulin
Numerous neurons in the brain are highly responsive to changes in the glucose concentration in their immediate environment. Specific neuron populations are either inhibited or stimulated by increases in glucose concentration. Many of these neurons express neuropeptides such as orexin, melanin-concentrating hormone (MCH), and neuropeptide Y, believed to be involved in the central regulation of energy balance by affecting food intake.
Insulin’s Role in Intracellular Potassium Homeostasis
Insulin plays a crucial role in maintaining intracellular potassium (K+) concentrations, a function that becomes particularly significant in insulin deficiency states such as diabetes mellitus. In the absence of insulin, K+ shifts from the cells into the extracellular fluid, and due to glucose-induced osmotic diuresis, there is an increased loss of K+ in the urine. This can result in a seemingly normal plasma K+ concentration when measured. However, when insulin replacement is initiated, a potentially harmful hypokalemia can develop as K+ rapidly re-enters the cells, especially when the body’s total K+ content is reduced

Mechanisms of insulin resistance in type 2 diabetes mellitus
Tissue | Pathophysiologic defect | Result |
Skeletal muscle | Impaired translocation of GLUT4 to the plasma membrane of myocytes. Defective tyrosine phosphorylation of various proximal insulin receptor substrates. | Reduction in glucose uptake by skeletal muscle[5]. |
Adipose tissue | Reduced sensitivity of adipose to tissue to the anti-lipolytic action of insulin (an unknown underlying mechanism) Release of adipokines associated with insulin resistance. | Increased lipolysis[39] |
Liver | Adipokines and free fatty acids (adipose tissue-derived) impair insulin signaling in the liver. | Impaired suppression of hepatic glucose output, which results in persistent hyperglycemia[40] |
References
1. Steiner, D.F., Park, S.-Y., Støy, J., Philipson, L.H., and Bell, G.I. (2009) A brief perspective on insulin production. Diabetes Obes. Metab., 11 Suppl 4, 189–196.2. Davidson, H.W. (2004) (Pro)Insulin processing: a historical perspective. Cell Biochem. Biophys., 40 (3 Suppl), 143–158.3. Fu, Z., Gilbert, E.R., and Liu, D. (2013) Regulation of Insulin Synthesis and Secretion and Pancreatic Beta-Cell Dysfunction in Diabetes. Curr. Diabetes Rev., 9 (1), 25–53.4. Norton, L., Shannon, C., Gastaldelli, A., and DeFronzo, R.A. (2022) Insulin: The master regulator of glucose metabolism. Metabolism., 129, 155142.5. Petersen, M.C., and Shulman, G.I. (2018) Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev., 98 (4), 2133–2223.6. Rorsman, P., Eliasson, L., Renström, E., Gromada, J., Barg, S., and Göpel, S. (2000) The Cell Physiology of Biphasic Insulin Secretion. Physiology, 15 (2), 72–77.7. Gerich, J.E. (2002) Is Reduced First-Phase Insulin Release the Earliest Detectable Abnormality in Individuals Destined to Develop Type 2 Diabetes? Diabetes, 51 (suppl_1), S117–S121.8. Meier, J.J. (2016) Chapter 32 – Insulin Secretion, in Endocrinology: Adult and Pediatric (Seventh Edition) (eds.Jameson, J.L., De Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., and Weir, G.C.), W.B. Saunders, Philadelphia, pp. 546-555.e5.9. Rorsman, P., and Ashcroft, F.M. (2018) Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev., 98 (1), 117–214.10. K, K., and P, N. (2014) Metabolic regulation of insulin secretion. Vitam. Horm., 95.11. Wiederkehr, A., and Wollheim, C.B. (2006) Minireview: implication of mitochondria in insulin secretion and action. Endocrinology, 147 (6), 2643–2649.12. Rorsman, P., and Braun, M. (2013) Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol., 75, 155–179.13. Taguchi, N., Aizawa, T., Sato, Y., Ishihara, F., and Hashizume, K. (1995) Mechanism of glucose-induced biphasic insulin release: physiological role of adenosine triphosphate-sensitive K+ channel-independent glucose action. Endocrinology, 136 (9), 3942–3948.14. Henquin, J.C. (2009) Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia, 52 (5), 739–751.15. Ježek, P., Jabůrek, M., Holendová, B., and Plecitá-Hlavatá, L. (2018) Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Mol. J. Synth. Chem. Nat. Prod. Chem., 23 (6), 1483.16. Ramos, P.A., Lytle, K.A., Delivanis, D., Nielsen, S., LeBrasseur, N.K., and Jensen, M.D. (2021) Insulin-Stimulated Muscle Glucose Uptake and Insulin Signaling in Lean and Obese Humans. J. Clin. Endocrinol. Metab., 106 (4), 1631–1646.17. Chang, L., Chiang, S.-H., and Saltiel, A.R. (2004) Insulin Signaling and the Regulation of Glucose Transport. Mol. Med., 10 (7), 65–71.18. Wasserman, D.H. (2022) Insulin, Muscle Glucose Uptake, and Hexokinase: Revisiting the Road Not Taken. Physiology, 37 (3), 115–127.19. Abdul-Ghani, M.A., and DeFronzo, R.A. (2010) Pathogenesis of Insulin Resistance in Skeletal Muscle. J. Biomed. Biotechnol., 2010, 476279.20. Petersen, M.C., Vatner, D.F., and Shulman, G.I. (2017) Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol., 13 (10), 572–587.21. Lewis, G.F., Carpentier, A.C., Pereira, S., Hahn, M., and Giacca, A. (2021) Direct and indirect control of hepatic glucose production by insulin. Cell Metab., 33 (4), 709–720.22. Boucher, J., Kleinridders, A., and Kahn, C.R. (2014) Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol., 6 (1), a009191.23. Belfiore, A., Malaguarnera, R., Vella, V., Lawrence, M.C., Sciacca, L., Frasca, F., Morrione, A., and Vigneri, R. (2017) Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev., 38 (5), 379–431.24. Bolaños, J.P., Almeida, A., and Moncada, S. (2010) Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci., 35 (3), 145–149.25. Cohen, P., Nimmo, H.G., and Proud, C.G. (1978) How does insulin stimulate glycogen synthesis? Biochem. Soc. Symp., (43), 69–95.26. Fang, X., Yu, S.X., Lu, Y., Bast, R.C., Woodgett, J.R., and Mills, G.B. (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. U. S. A., 97 (22), 11960–11965.27. Patel, S., Doble, B., and Woodgett, J.R. (2004) Glycogen Synthase Kinase-3 in Insulin and Wnt Signalling : a Double-edged Sword? Biochem. Soc. Trans., 32 (Pt 5), 803–808.28. Adeva-Andany, M.M., González-Lucán, M., Donapetry-García, C., Fernández-Fernández, C., and Ameneiros-Rodríguez, E. (2016) Glycogen metabolism in humans. BBA Clin., 5, 85–100.29. Vallon, V., and Thomson, S.C. (2017) Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia, 60 (2), 215–225.30. Mergenthaler, P., Lindauer, U., Dienel, G.A., and Meisel, A. (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci., 36 (10), 587–597.31. (2021) Gluconeogenesis- Reaction and regulation.32. Sanvictores, T., Casale, J., and Huecker, M.R. (2023) Physiology, Fasting, in StatPearls, StatPearls Publishing, Treasure Island (FL).33. Hatting, M., Tavares, C.D.J., Sharabi, K., Rines, A.K., and Puigserver, P. (2018) Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci., 1411 (1), 21–35.34. Gerich, J.E., Meyer, C., Woerle, H.J., and Stumvoll, M. (2001) Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care, 24 (2), 382–391.35. Hanson, R.W., and Owen, O.E. (2013) Gluconeogenesis, in Encyclopedia of Biological Chemistry (Second Edition) (eds.Lennarz, W.J., and Lane, M.D.), Academic Press, Waltham, pp. 381–386.36. Martin-Requero, A., Ayuso, M.S., and Parrilla, R. (1986) Rate-limiting steps for hepatic gluconeogenesis. Mechanism of oxamate inhibition of mitochondrial pyruvate metabolism. J. Biol. Chem., 261 (30), 13973–13978.37. Pilkis, S.J., and Granner, D.K. (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu. Rev. Physiol., 54, 885–909.38. van Schaftingen, E., and Gerin, I. (2002) The glucose-6-phosphatase system. Biochem. J., 362 (Pt 3), 513–532.39. Morigny, P., Houssier, M., Mouisel, E., and Langin, D. (2016) Adipocyte lipolysis and insulin resistance. Biochimie, 125, 259–266.40. Meshkani, R., and Adeli, K. (2009) Hepatic insulin resistance, metabolic syndrome and cardiovascular disease. Clin. Biochem., 42 (13), 1331–1346.