Acromegaly and Gigantism. Rooted in the excess of growth hormone, these two conditions, while closely related, manifest in distinct ways and present unique challenges to those they affect. In this article, we dive deep into the realms of Acromegaly and Gigantism, exploring their nuances, from their pathophysiology to their clinical presentation.
Pituitary Gigantism: Overview and Management
Pituitary gigantism occurs in children or adolescents when a growth hormone (GH)-secreting pituitary tumor develops before the fusion of the epiphyseal growth plates. In contrast, GH-secreting tumors in adults, where epiphyseal fusion is complete, lead to acromegaly, marked by acral changes rather than linear growth.
Pituitary gigantism is a rare condition. In extreme cases starting in infancy, it can result in extraordinary height, with the tallest recorded individual reaching 8 ft 11 in (272 cm). Typically, untreated pituitary giants exceed 7 ft in height. These GH-secreting tumors are often sporadic but can also be part of syndromes like Multiple Endocrine Neoplasia type 1, McCune-Albright syndrome, and the Carney complex.
Besides pituitary adenomas, gigantism may also arise from ectopic tumors secreting GH-releasing hormone (GHRH) or due to hypothalamic dysfunction. Along with accelerated growth, patients may develop features seen in acromegaly, including soft tissue overgrowth, dental malocclusion, a low-pitched voice, headaches, hyperhidrosis, and complications like diabetes mellitus, hypertension, obstructive sleep apnea, and cardiac dysfunction. The mass effects of large GH-producing pituitary macroadenomas include visual field defects, oculomotor pareses, and pituitary insufficiency.
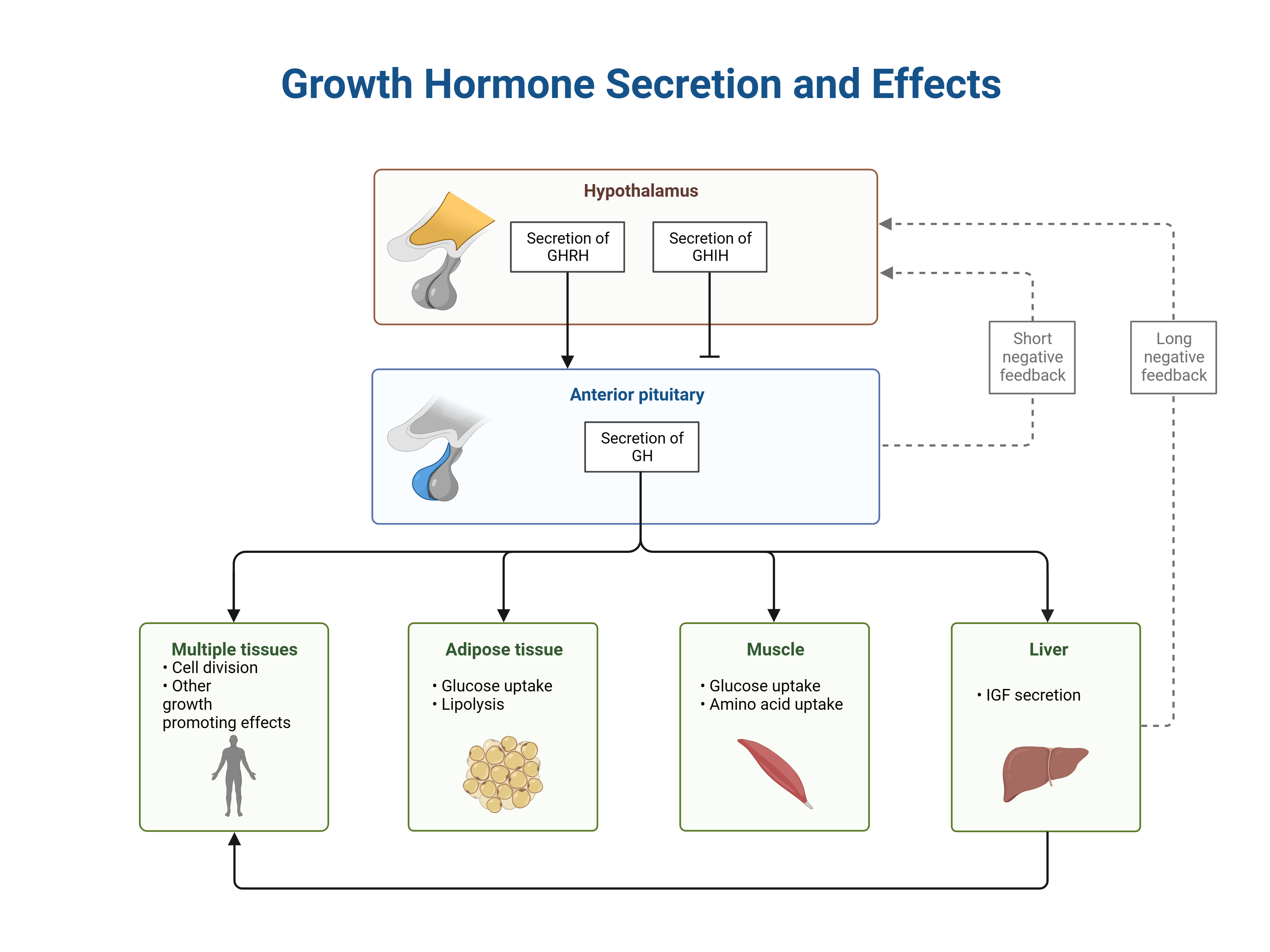
However, it’s important to recognize that most children with accelerated growth do not have pituitary gigantism. Conditions like precocious puberty, genetic tall stature, and hyperthyroidism are more common causes. The diagnosis involves ruling out other causes of rapid growth and confirming through biochemical tests, including non-suppression of GH after an oral glucose load and increased serum levels of insulinlike growth factor 1 (IGF-1). Magnetic Resonance Imaging (MRI) of the pituitary and visual field examination are also crucial for diagnosis.
Treatment for pituitary gigantism aims to control GH excess, remove the tumor, and preserve normal pituitary function. The primary treatment is transsphenoidal adenectomy, often supplemented by Gamma knife radiotherapy and pharmacotherapy. Early intervention is essential to mitigate the long-term effects of excessive GH and to address the mass effects of the tumor.
Acromegaly: Clinical Features and Management
Acromegaly is a syndrome resulting from chronic excess of growth hormone (GH), typically due to a GH-secreting pituitary tumor. Described by Pierre Marie in 1886, acromegaly leads to distinctive physical changes and several health complications if left untreated. Unlike pituitary gigantism, which occurs in children and adolescents before the closure of epiphyseal plates, acromegaly occurs in adults and is characterized by acral and soft tissue overgrowth, progressive dental malocclusion, degenerative arthritis, a low-pitched voice, headaches, hyperhidrosis, oily skin, nerve entrapment (such as carpal tunnel syndrome), muscle weakness, carbohydrate intolerance, hypertension, and cardiac dysfunction.
Patients with acromegaly often exhibit characteristic facial changes, including coarsening of facial features, prognathism, frontal bossing, spadelike hands, and wide feet. There’s often a history of progressive increase in shoe, glove, ring, or hat size. These changes can be gradual and might go unrecognized for years, with an average diagnosis delay of 8.5 years from the onset of symptoms.
Diagnosing Acromegaly involves more than just high plasma GH levels, as these can be elevated in other conditions like diabetes mellitus, hepatic or renal failure, and protein-calorie malnutrition. The definitive diagnosis is based on two criteria: failure to suppress GH to less than 0.4 ng/dL after an oral glucose load and elevated serum insulinlike growth factor 1 (IGF-1) levels. Magnetic Resonance Imaging (MRI) of the pituitary and visual field examination are important diagnostic tools. If no adenoma is detected, plasma GH-releasing hormone (GHRH) concentrations and CT scans of the chest and abdomen may be performed to look for ectopic GHRH-producing tumors.
The treatment of acromegaly aims to mitigate the long-term effects of GH excess, remove the sellar mass, and preserve normal pituitary function. Transsphenoidal adenectomy by an experienced neurosurgeon is the preferred treatment, often supplemented with Gamma knife radiotherapy and pharmacotherapy. Post-surgery, there is significant regression of soft tissue excess, but bone changes are permanent. Follow-up treatments might include oral and plastic surgery for residual soft tissue and bone abnormalities, and joint replacement for disabling osteoarthropathy. Regular colonoscopic screenings are advised due to the increased risk of colorectal cancer.
Comparison of acromegaly and gigantism
| Factor | Gigantism | Acromegaly |
| Definition | Excessive growth hormone (GH) production in children or adolescents before epiphyseal plate closure. | Excessive GH production in adults after epiphyseal plate closure. |
| Cause | GH-secreting pituitary tumor. | GH-secreting pituitary tumor. |
| Age of Onset | Childhood or adolescence. | Adulthood. |
| Primary Manifestation | Excessive linear growth, leading to extreme height. | Enlargement of acral parts (hands, feet), and soft tissue overgrowth. |
| Typical Height | Over 7 feet, can be extreme in cases starting in infancy. | Normal adult height with enlarged extremities. |
| Associated Features | Accelerated growth rate, may have symptoms similar to acromegaly. | Coarsening of facial features, prognathism, frontal bossing, soft tissue excess, potential for joint problems, and systemic complications. |
| Complications | Early onset of complications associated with excessive GH (e.g., diabetes, hypertension). | Carpal tunnel syndrome, diabetes mellitus, hypertension, obstructive sleep apnea, and cardiac dysfunction. |
| Treatment | Surgery to remove the tumor, hormone therapy. | Surgery (transsphenoidal adenectomy), radiotherapy, pharmacotherapy. |
| Diagnosis | Elevated GH and insulin-like growth factor 1 (IGF-1) levels, MRI for tumor detection. | Elevated GH not suppressed by glucose, increased IGF-1, MRI for tumor detection. |
| Prognosis | Dependent on early diagnosis and treatment efficacy. Bone changes may be permanent. | Soft tissue changes can regress post-treatment, but bone changes are permanent. Regular monitoring required for associated complications. |
Quiz
Question 1:
A 15-year-old boy presents with a significant increase in height over the past year, now towering over his peers at 6 feet 5 inches. He also complains of joint pains and headaches. His parents mention a remarkable increase in his shoe size. Physical examination reveals large hands and feet and a slightly protruded jaw. Blood tests show elevated levels of growth hormone and insulin-like growth factor 1 (IGF-1). An MRI of the brain indicates a pituitary adenoma. What is the most likely diagnosis?
A) Acromegaly
B) Gigantism
C) Thyroid storm
D) Cushing’s syndrome
Answer:
B) Gigantism
Explanation:
The correct answer is Gigantism. This condition is characterized by excessive growth hormone production in children or adolescents before the closure of their epiphyseal growth plates, leading to abnormal increases in height and overgrowth of extremities. The patient’s significant increase in height, large hands and feet, and elevated GH and IGF-1 levels, along with the presence of a pituitary adenoma, are classic indicators of gigantism. Acromegaly, choice A, occurs in adults after epiphyseal plate closure and typically doesn’t result in increased height, but rather in acral enlargement and soft tissue overgrowth. Thyroid storm (C) and Cushing’s syndrome (D) present with different clinical features and are not associated with the presented symptoms.
Question 2:
A 45-year-old woman visits your clinic with complaints of a gradual change in her facial appearance, including a more prominent jaw, enlarged lips, and wider nose. She also reports having joint pains and a recent increase in her ring and shoe sizes. Lab tests reveal elevated serum growth hormone levels that do not adequately suppress following an oral glucose tolerance test. MRI of the pituitary gland shows a 1.5 cm adenoma. What is the most likely diagnosis?
A) Gigantism
B) Acromegaly
C) Hyperthyroidism
D) Addison’s disease
Answer:
B) Acromegaly
Explanation:
The correct diagnosis is Acromegaly. This condition is characterized by the excess production of growth hormone in adults, leading to the enlargement of facial features, extremities, and soft tissues. The patient’s symptoms of gradual facial changes, joint pains, increased ring and shoe sizes, elevated growth hormone levels, and a pituitary adenoma on MRI are typical of acromegaly. Gigantism (A) presents similarly but occurs in children and adolescents and results in increased height. Hyperthyroidism (C) and Addison’s disease (D) have different symptom profiles and do not lead to the overgrowth of facial features or extremities.