
Pancreas-related physical examination Signs
Diabetes mellitus
Acanthosis nigricans
Acanthosis nigricans (AN) is a hyperpigmented lesion with a predilection for flexural areas such as the neck, axilla and intertriginous regions of the body. Patients with type 2 diabetes mellitus, an obese phenotype, or the metabolic syndrome are predisposed to developing this classic skin lesion.
Other endocrinopathies associated with acanthosis nigricans
- Cushing’s syndrome
- Addison’s disease
- PCOS
- CAH
- Hyperandrogenism insulin resistance acanthosis nigricans (HAIR-AN) syndrome
- Acromegaly
Diabetic dermopathy
Diabetic dermopathy (DD) is reported as a cardinal skin manifestation of both type 1 and type 2 diabetes mellitus. It presents as hyperpigmented and circumscribed macules noted over the pretibial skin. These characteristic lesions have a predilection for areas prone to trauma and are present in up to 55% of patients with diabetes mellitus (DM)
Acrochordons (skin tags)
Skin tags have a reported prevalence of 25% in patients with type 2 diabetes mellitus. The lesions are pedunculated cutaneous fibromas distributed over flexural areas in the neck and axilla, in a pattern akin to the distribution of AN
Necrobiosis lipoidica diabeticorum
Necrobiosis lipoidica diabeticorum (NLD) is a rare clinical finding in patients with either type 1 or type 2 diabetes mellitus. The lesions typically start as a red-brown papule that progressively increases in diameter and eventually changes into the classic waxy, yellowish lesion. There is an occasional central ulceration, which manifests as an atrophic center.
Lipodystrophy due to insulin injections
Localized lipodystrophy due to insulin injections is an umbrella term that consists of the lipoatrophy (LA) and lipohypertrophy (LH) subtypes
LA tends to appear as an area of subcutaneous tissue loss, which creates a dimple in the skin. LH, however, has a firm, rubbery consistency that is palpable in the subcutaneous tissue plane. Occasionally, LH lesions may be soft, making them difficult to discern on routine physical examination. Ultrasound is more sensitive than the clinical exam for the detection of lipodystrophy.
Lipodystrophic areas can lead to malabsorption of insulin (either delayed or more rapid) from the subcutaneous tissue. This reportedly causes wide variability in glycemic control and can drastically impact patient care if it remains undiagnosed.
In a meta-analysis of over 12,000 patients with diabetes mellitus, the pooled prevalence estimate of lipodystrophy was astonishingly high at 38% (95% confidence interval 29-46%). LH is best appreciated clinically by palpating the affected area with the pulp of the fingertips.
Other skin manifestations of diabetes mellitus
| Lesion | Clinical findings | Pathophysiology |
| Eruptive xanthoma | Yellowish papules located over the trunk and extensor surfaces such as the elbows and knees. | Reduced lipoprotein lipase activity due to insulin resistance or insulinopenia causes impaired storage of triglycerides in adipose tissue. Following which excess circulating triglycerides accumulate in the skin. |
| Bullosis Diabeticorum | Usually located on the lower extremities. Bullae tend to contain a clear fluid which can be aspirated when it causes discomfort. | unknown |
| Scleroderma diabeticorum | Thick, indurated plaques tend to be distributed over the neck and upper back. | Reduced breakdown of collagen fibers due to nonenzymatic glycosylation of dermal collagen |
| Granuloma annulare | Firm, erythematous papules with a ring-like configuration. | Microangiopathy, lymphocytic infiltration, and eventual connective tissue degeneration |
Diabetic retinopathy on direct ophthalmoscopy
The direct ophthalmoscope, an essential component of the physical exam, is increasingly not being utilized by present-day physicians. Recent evidence points to an increasing trend of low competency in the use of the direct ophthalmoscope.
Nonetheless, direct ophthalmoscopy conducted by an ophthalmologist has high specificity but a low sensitivity of 34-50% in detecting early diabetic retinopathy
There are two forms of diabetic retinopathy. Non-proliferative diabetic retinopathy (NPDR) consists of microaneurysms, dot “hemorrhages,” cotton wool spots, and hard exudates. Dot hemorrhages are microaneurysms seen in cross-section.
Proliferative diabetic retinopathy (PDR) has neovascularization, scar tissue formation with or without vitreous hemorrhage or retinal detachment as manifestations
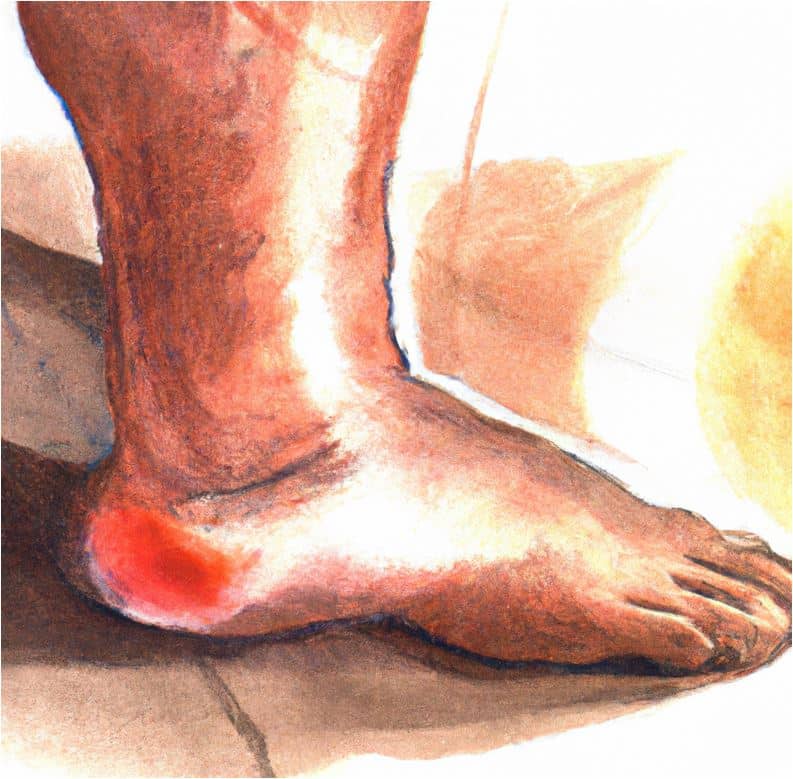
The diabetic foot
The dreaded diabetic foot, a complication of uncontrolled diabetes predisposes patients to foot ulcers, with an estimated lifetime risk of 25%. There are two reported variants of the diabetic foot in the literature; these include the neuropathic and neuroischemic subtypes.
As the name implies, the neuropathic foot has neuropathy as the underlying microvascular complication. The neuroischemic foot has neuropathy and vasculopathy co-existing in the same foot.
Rabson-Mendenhall Syndrome (Type A Insulin Resistance)
Acanthosis nigricans
Acanthosis nigricans is a dermatologic manifestation of type A insulin resistance syndromes. The clinical features of acanthosis nigricans have been previously described.
Other physical manifestations of RMS and their underlying pathophysiologic mechanisms
| Physical finding(s) | Pathophysiology |
| Organomegaly (phallic enlargement, clitoromegaly, and nephromegaly) | IGF-1, which is a growth factor, shares structural homology with insulin. Hyperinsulinemia accounts for organomegaly due to the effects of insulin on the IGF-1 receptor |
| Hypertrichosis and xerosis | Insulin binding to IGF-1 receptors in the integument |
Glucagonoma
Necrolytic migratory erythema
Necrolytic migratory erythema (NME) is a pathognomonic dermatosis of the glucagonoma syndrome. NME is the presenting feature of the glucagonoma syndrome in up to 70% of patients. Skin lesions are annular, crusted, erythematous plaques distributed over mainly intertriginous areas, but may be seen on the extremities and trunk as well. The lesions may be easily misdiagnosed as other causes of dermatoses, leading to delayed diagnosis in most patients.
What are some other clinical features of glucagonoma syndrome?
Clinical features and underlying mechanisms of some manifestations of the glucagonoma syndrome
| Clinical feature | Mechanism(s) |
| Weight loss | Glucagon acts on Glucagon-like (GLP-1) receptors in central satiety centers (anorexigenic pathway) to facilitate weight loss. Stimulates energy expenditure by activating thermogenesis in brown adipose tissue. |
| Cardiomyopathy and heart failure | Glucagon binding to its G-protein coupled receptor increases intracellular cyclic AMP, which activates protein kinase A (PKA). PKA mediates the phosphorylation of L-type calcium channels of the cardiomyocyte sarcolemma. This results in an influx of calcium, which promotes increased myocardial contraction. Prolonged activation of the sarcoplasmic reticulum leads to calcium leak, accentuated myocardial contractility, and eventual cardiac remodeling. |
What is pseudoglucagonoma syndrome, and how can it be differentiated from classic glucagonoma syndrome?
Pseudoglucagonoma syndrome can present with clinical features of a glucagonoma in the absence of an alpha cell tumor of the pancreas. Patients may have either elevated or normal glucagon levels and, in some cases, present with the characteristic NME rash. Malignancies, chronic liver disease, pancreatitis, and non-tropical sprue are known causes of pseudoglucagonoma syndrome.
Carcinoid Syndrome
Cutaneous flushing
Cutaneous flushing has a mean prevalence of 78%, based on cumulative evidence from case series. It classically involves the face, neck, and upper chest and is usually triggered by amine-rich foods, pharmacologic agents, or emotional stress.
Repeated episodes of flushing which tend to be transient (lasting 10-30 minutes) are characteristic of midgut carcinoids. A longer-lasting duration of flushing (up to several hours) is more characteristic of foregut carcinoids.
Associated endocrinopathies/conditions that can present with flushing
Pheochromocytomas, Cushing’s syndrome, medullary thyroid cancer, rosacea, and mastocytosis.
Diarrhea
There is a variable reported prevalence of diarrhea, ranging from 58-100% with a mean of 78% in multiple case series. Diarrhea tends to be secretory and, as such, persists even when the patient is fasting. This should be contrasted from non-secretory diarrhea, which improves in the setting of a fast and is usually of other etiology.
Bronchospasm
Asthma-like symptoms present with audible wheezing and confirmatory rhonchi on auscultation. The prevalence of asthma-like features is, however, less frequent when compared to diarrhea and cutaneous flushing.
The reported prevalence rate based on large case series is about 3-18%. Carcinoid syndrome can be misdiagnosed as asthma, and treatment with standard anti-bronchospastic therapies can result in the worsening of symptoms.
Cardiac valvular lesions
Carcinoid heart disease(CHD), has a prevalence rate ranging from 11-70%. Patients can present with right-sided murmurs (tricuspid and pulmonary valves), peripheral edema, ascites, and other signs of right-sided heart failure.
Other cutaneous manifestations (Pellagra)
Pellagra is described in many medical texts as a syndrome composed of dermatitis, diarrhea, dementia, and death. The mnemonic “4Ds”, is well known by many medical professionals. It is a photosensitive rash that tends to involve sun-exposed areas such as the face, neck, and forearms. The rash is characteristically hyperpigmented and scaly.
VIPoma
Dehydration due to extrarenal losses
Secretory diarrhea is a component of WDHA (watery diarrhea, hypokalemia, hypochlorhydria or achlorhydria) syndrome, a common tetrad seen in patients with VIPomas. Due to the significant amount of gastrointestinal fluid and electrolyte losses, patients can develop dehydration with resultant hypotension and acute kidney injury.

Lipid disorders physical examination signs
Familial Hyperlipidemia Syndromes
Corneal arcus
Corneal arcus (CA) manifests clinically as a circumferential deposition of lipids in the cornea and appears as whitish-gray deposits around the cornea with a predilection for the superior and inferior limbus of the cornea. It eventually progresses to involve the entire peripheral rim of the cornea.
CA is widely accepted as a typical sign of the aging process, although it might signify the presence of dyslipidemia. A population-based prospective study involving more than 3600 subjects in Asia, assessed the association of CA with cardiovascular disease (CVD) events.
The authors prospectively evaluated CVD events after an initial baseline confirmation of the presence of CA. After adjusting for baseline traditional risk factors, CA was associated with a clinically significant increased odds ratio (OR) of 1.52 [1.07 to 2.16] for incident CVD.
Xanthomata and xanthelasma.
Xanthomas represent localized regions of accumulated lipids in the skin, subcutaneous tissues, and tendons. They may be plaque-like, papular, or nodular lesions, depending on the morphologic subtype.
Tuberous xanthomas are characteristically distributed over pressure points (sites of trauma) such as the elbows and knees. They are firm, yellowish-red papulonodular skin lesions, which vary in size from a few millimeters to massive xanthomas. Tuberous xanthomas occur in patients with familial hypercholesterolemia (Type IIa hyperlipoproteinemia) and familial dysbetalipoproteinemia (type III hyperlipoproteinemia).
Eruptive xanthomas (EX), on the other hand, tend to present in patients with Type I, IV, or V hyperlipoproteinemia. EX has a reported prevalence of 1.7 for every 10,000 patients. Secondary causes of EX include diabetes mellitus, hypothyroidism, alcohol use, cholestasis, and estrogens.
- They may vary from yellowish-orange to reddish-brown papules with a circumferential erythematous hue. EX are usually located over extensor surfaces of the extremities but may occur in flexural areas and the trunk.
- EX lesions exhibit the Koebner phenomenon, i.e., new lesions develop along the sites of cutaneous trauma or irritation[19].
Palmar xanthomas (PX) appear as yellowish-orange macules with a predilection for the palmar creases. They are pathognomonic for Type III hyperlipoproteinemia (familial dysbetalipoproteinemia) and may, on rare occasions, occur in patients with familial hypercholesterolemia. PX should be distinguished from planar xanthomas, which are whitish plaques and tend to extend beyond the confines of the palmar creases. Palmar xanthomas involving the palmar creases are classically referred to as xanthoma striatum palmare.
Xanthelasmas are the most common cutaneous xanthoma and are distributed over the inner canthus of the upper eyelids. The lesions are soft, yellowish plaques and may be seen in other sites apart from the periorbital region, such as the axilla, neck, and trunk. In a large prospective cohort study involving more than 12,000 subjects, investigators were able to demonstrate that xanthelasmas were independently associated with ischemic heart disease after controlling for some CVD risk factors such as elevated plasma cholesterol and triglycerides.
Skin crease (Frank’s sign)
Frank’s sign is a diagonal wrinkle involving the skin of the ear lobe. This presumed dermatologic manifestation of atherosclerotic disease was first reported by Dr. Sanders Frank in 1973, in a letter titled “Aural Sign of Coronary-Artery Disease,” published in the New Journal of Medicine (NEJM).
It is thought to increase with age and is associated with some cardiovascular risk factors. There is conflicting evidence in the medical literature regarding the association of Frank’s sign with atherosclerotic disease. The association of Frank’s sign with atherosclerotic disease might, however, not necessarily imply a direct causal relationship.
Lipemia retinalis
It is a rare fundoscopic examination finding in patients with markedly elevated serum triglycerides. The vessels in the posterior pole and peripheral aspect of the retina develop a creamy-white appearance that can be visualized on the fundoscopic exam. Some patients may have deterioration of visual acuity, which improves upon initiation of anti-lipidemic therapy.
Fredrickson classification of hyperlipoproteinemias
| Type | Primary hyperlipidemia classification | Elevated lipoprotein |
| I | Familial hyperchylomicronemia syndrome | Chylomicrons |
| IIa | Familial hypercholesterolemia | LDL |
| IIb | Familial combined hypercholesterolemia | LDL, VLDL |
| III | Familial dysbetalipoproteinemia | IDL |
| IV | Simple hypertriglyceridemia | VLDL |
| V | Familial hypertriglyceridemia | Chylomicrons, VLDL |
Congenital Leptin Deficiency
Obesity
Congenital leptin deficiency is associated with significant early-onset obesity. Patients exhibit food-seeking behavior and present with extreme obesity at a very young age
Questions you might be asked on clinical rounds
What is the genetic basis of congenital leptin deficiency?
The majority of patients with congenital leptin deficiency were conceived in consanguineous relationships. It occurs as a result of a genetic mutation in the leptin receptor (LEPR) gene; thus, a defective protein is translated and transcribed.
What are the other endocrine effects of leptin?
- Hypothalamic-pituitary-thyroidal axis: Leptin regulates the release of thyroid-stimulating hormone (TSH). Central hypothyroidism occurs due to leptin-mediated signaling defects in the hypothalamic-pituitary-thyroid axis. Indeed, the amelioration of hypothyroidism occurs after optimal leptin replacement therapy.
- Hypogonadotropic-pituitary-gonadal axis: Leptin regulates the release of gonadotropin-releasing hormone (GnRH). Patients with congenital leptin deficiency are hypogonadal due to defective GnRH release.
Proopiomelanocortin (POMC) Deficiency
Triad of Obesity, adrenal insufficiency, and reddish hair
Early-onset obesity was reported as a clinical finding in the first case of POMC deficiency. Other clinical findings in this recently discovered monogenic cause of obesity included reddish pigmentation of body hair and features of central adrenal insufficiency. Multiple case reports have been published since the index case.
What are the other clinical features seen in patients with POMC deficiency?
Clinical features of POMC deficiency and their underlying mechanisms
| Clinical features | Mechanism |
| Hypopigmented skin | Defects in MSH mediated melanocyte activation |
| Hypogonadotropic Hypogonadism. | Defects in POMC mediated signaling of GnRH release |
| Central hypothyroidism | Defects in POMC mediated signaling of TRH release |
| Hyponatremia | Increased ADH secretion in the setting of central hypocortisolemia. Elevated CRH is a secretagogue for antidiuretic hormone |
CRH Corticotropin-releasing hormone
Adapted from Çetinkaya et al. (2018) A Patient with Proopiomelanocortin Deficiency: An Increasingly Important Diagnosis to Make. J Clin Res Pediatr Endocrinol 10:68–73
Lipodystrophy Syndromes
Atrophy of adipose tissue
The distribution of adipose tissue atrophy is variable in patients with lipodystrophy syndromes and is dependent on the underlying cause
Distribution of fat loss in lipodystrophy syndromes
| Type of lipodystrophy | Distribution of adipose atrophy |
| Familial partial lipodystrophy (Dunnigan variety) | Loss of subcutaneous tissue fat in the extremities and trunk. Increased fat deposition in the supraclavicular region |
| Acquired generalized lipodystrophy | A generalized progressive loss of subcutaneous tissue fat |
| Acquired partial lipodystrophy (APL, Barraquer-Simons syndrome) | Selective loss of subcutaneous tissue fat involving the trunk, upper limbs, and head |
| Localized lipodystrophy | Medication-induced loss of subcutaneous tissue. It tends to involve sites of injection e.g. insulin-mediated lipodystrophy |
Hepatomegaly
The rate of splenomegaly varies from as low as 29% in acquired partial lipodystrophy to as high as 84% in congenital generalized lipodystrophy
Mechanisms underlying other manifestations of lipodystrophy syndromes
| Clinical feature | Mechanism |
| Acanthosis nigricans | Insulin resistance |
| Virilization | Hyperinsulinemia induced hyperandrogenemia |
| Xanthomas | Hypertriglyceridemia-induced |
| Prominent veins and muscles in the extremities | The loss of subcutaneous tissue fat makes superficial vessels more visible |
Calcium and Parathyroid physical examination signs
Primary Hyperparathyroidism
Acute abdomen
Hyperparathyroidism can cause a myriad of nonspecific abdominal complaints and may sometimes present as an acute abdomen. Acute pancreatitis and complications of hypergastrinemia, e.g., peptic ulcer disease, may result in an acute surgical abdomen. Acute pancreatitis has been associated with hyperparathyroidism since it was first described in the 1950s. A study in 2006 reported a 28 fold increase in the risk of pancreatitis among patients with hyperparathyroidism when compared to controls in the general population.
Fragility fractures
Fragility fractures due to hyperparathyroidism in young subjects have been reported. The loss of cortical bone mineral density is disproportionately higher than that of lamellar (cancellous) bone, making the distal third radius, an ideal site to evaluate with bone densitometry in patients with this condition.
Band keratopathy/cataracts
Band keratopathy is a clinical finding in various conditions, including hyperparathyroidism. It is a whitish-grey opacification involving the cornea with a predilection for the nasal or temporal regions of the cornea
Hypertension
Hypertension has been associated with primary hyperparathyroidism for several decades. There is a widely reported prevalence of 20-80%, most likely due to study heterogeneity. A large retrospective study involving more than 4000 subjects reported higher all-cause mortality and cardiovascular-specific deaths when patients with primary hyperparathyroidism were compared to matched controls.
Hypoparathyroidism
Trousseau’s sign and Chvostek sign in the setting of hypocalcemia
Trousseau’s sign usually manifests as limb spasms, best elicited by placing the cuff of a sphygmomanometer over the upper arm and inflating it to at least 20mmHg above the systolic blood pressure. It is both sensitive and specific for clinically significant hypocalcemia.
A study reported the prevalence of Trousseau’s sign as being up to 94% in patients with biochemically confirmed hypocalcemia, compared with 1% in patients with normal serum calcium.
Dr. Franz Chvostek first reported a case of latent tetany in 1870. Chvostek’s sign is a unilateral twitching of the facial musculature due to the tapping of the superficial part of the facial nerve. Percussion of the facial nerve can either be done anterior to the external acoustic meatus or directly on the cheek.
In contrast to Trousseau’s sign, Chvostek’s sign has poor specificity and sensitivity for hypocalcemia. A systematic review reported the sensitivity of Chvostek’s sign as ranging from 0% to 100 % with a specificity of 78.8% to 100% among patients with hypocalcemia.
Seizures
Hypocalcemia-induced seizures can be a presentation of hypoparathyroidism, although any perturbation in calcium homeostasis, irrespective of the underlying cause, can result in seizures.
Hypotension
A case of hypocalcemia-induced hypotension was reported in a letter published in the Journal of the American Medical Association (JAMA) in 1972. The patient had refractory hypotension in the setting of uremic pericardial effusion and clinically symptomatic hypocalcemia.
Interestingly, the correction of hypocalcemia led to prompt resolution of hypotension before the performance of pericardiocentesis. There have been other reports of hypocalcemia-induced hypotension since then.
Papilledema
Papilledema, a rare fundoscopic finding in patients with severe hypocalcemia, is characterized by a blurring of the optic disk margin.
Pseudohypoparathyroidism
Short Stature
Short stature is a cardinal clinical finding in Albright hereditary osteodystrophy (AHO). The classic AHO phenotype is characterized by round facies, obesity, brachydactyly, and short stature.
Obesity
Most adults with PHP1A have a body mass index (BMI) >25kg/m2. The reported prevalence of obesity is more than 66%, higher than the 32% prevalence rate reported for the general population.
Brachydactyly
The characteristic skeletal feature of PHP happens to be shortening of the metacarpals and metatarsals. This skeletal change tends to involve the fourth and fifth metacarpals and metatarsals
Dental manifestations.
Multiple dental abnormalities including delayed or even failed eruption of the teeth, blunting of the roots, hypodontia, and ankylosis. Patients should be referred to dentists for optimal care of the teeth.
Clinical and biochemical features of some forms of iPPSD
| iPPSD | AHO | Other hormone Resistance States. | PTH resistance |
| PPHP | Present | Absent | Absent |
| PHP1A | Present | Present | Present |
| PHP1B | Absent | Infrequent | Present |
| PHP1C | Present | Present | Present |
iPPSD inactivating PTH/PTHrp signaling disorders, AHO Albright’s hereditary osteodystrophy.
Other hormone resistance states LH and TSH resistance
PTH resistance low calcium, high phosphorus, and a paradoxically high PTH
PHP1C Pseudohypoparathyroidism type 1C
Questions you might be asked on clinical rounds
What is the classic phenotype of AHO?
Dr. Fuller Albright first described Albright hereditary osteodystrophy (AHO) in 1942. The classic phenotype has the following features, brachydactyly, short stature, and round facies. The classification system for this group of inactivating PTH/PTHrp signaling disorders is based on the presence or absence of the classic AHO phenotype and Gsɑ activity in response to exogenous PTH administration.
What are the other endocrinopathies which might be associated with some forms of pseudohypoparathyroidism?
- Resistance to the effects of TSH at the level of the thyroid gland results in hypothyroidism
- Gonadotropin resistance leading to delayed puberty, oligomenorrhea, and cryptorchidism
- Growth hormone-releasing hormone (GHRH) resistance causes GH deficiency.
- Prolactin deficiency
Paget’s Disease of Bone
Fractures and bone deformity
Patients with Paget’s disease of bone (PDB) have a significant clinical fracture prevalence ranging between 10 to 30%. They may either be incomplete fissure fractures involving part of the cortical bone or complete transverse fractures. Fractures typically affect weight-bearing bones, which have a pre-existing deformity. Bowing deformities tend to involve weight-bearing long bones in the lower extremity. Patients may also present with skull and facial deformities such as frontal bossing.
Congestive heart failure
Heart failure can occur in patients with extensive pagetoid changes in the bone; it is, however, an uncommon presentation of PDB.
Sensorineural hearing loss
Sensorineural hearing loss (SNHL) is a known neurologic complication of PDB involving the skull. The classic Weber and Rinne tests, which involve the use of a tuning fork can be used at the bedside to differentiate conductive from SNHL. The tests require the use of a 512Hz (Hertz) tuning fork in assessing both bone and air conduction.
A recent systematic review evaluated the accuracy of the Weber and Rinne tests. It showed wide variability in the sensitivity and specificity of these tests since adherence to testing protocols were operator-dependent. We will refer readers to other clinical examination texts for the standard protocol of these tuning fork tests.
Interestingly it is reported that Ludwig van Beethoven suffered from PDB and that his SNHL may have influenced some of his musical compositions.
Questions you might be asked on clinical rounds
What are the other cardiac manifestations of Paget’s disease, apart from high output cardiac failure?
Aortic stenosis, atherosclerosis, and endocardial calcifications
How are patients with PGD more likely to present?
Up to 70% of patients are asymptomatic due to a prolonged period of latency in PGD. In symptomatic patients, a dull and deep aching pain involving pagetic bones is a more likely complaint at presentation.
Hereditary Vitamin D Resistant Rickets Type 2 (HVDRR-II)
Rickets
Dr. Fuller Albright reported a case of vitamin D resistant rickets in 1937. He postulated the underlying cause of rickets in his case report as being due to “an intrinsic resistance to the anti-rachitic action of vitamin D”. The clinical features of rickets occur at a young age and classically manifest before fusion of the growth plates.
Rickets tends to affect the distal forearms, knees, and costochondral regions. The typical features include swelling of the costochondral joints (rachitic rosary), widening of the wrist joint, and an exaggerated genu varum in children.
X-Linked Hypophosphatemic Rickets
Short Stature
Patients with X-linked Hypophosphatemic Rickets (XLHR) have short stature, with the length of the lower limbs more significantly affected than that of the trunk. This results in an abnormal upper segment to lower segment ratio (lower segment = from the top of the symphysis pubis to the heel; upper segment = height minus lower segment). Early treatment with oral phosphate supplementation and calcitriol improves growth rates and other skeletal outcomes
Dental abscess
Unprovoked dental abscesses occur in the absence of trauma or dental caries. Active dental surveillance to optimize oral hygiene is recommended, and there are no active therapies to prevent this complication.
What is the underlying cause of X-linked hypophosphatemia (XLH)?
A mutation in the PHEX (phosphate-regulating gene with homologies to endopeptidase on the X chromosome) gene results in impaired expression of a peptidase involved in the inactivation of FGF-23. This results in increased circulating levels of FGF-23, which causes hypophosphatemia and reduced bone mineralization.
There is a new FDA approved monoclonal antibody for the treatment of XLH called Burosumab. Burosumab binds circulating intact FGF23 and, thereby, blocks its biologic effects in target tissues. It improves renal tubular phosphate conservation, serum phosphorus levels, and promotes linear growth.
An important differential diagnosis of XLH is Tumor-induced osteomalacia (TIO). TIO is caused by increased production of FGF-23 by mesenchymal tumors. FGF-23 causes reduced insertion of sodium phosphate channels in the renal tubules, which results in increased urinary loss of phosphorus.
Besides, there is decreased production of calcitriol due to FGF-23 mediated inactivation of 1ɑ hydroxylase activity. Additionally, affected patients develop poor bone mineralization due to significant hypophosphatemia.
Reproduction related physical examination signs
Turner Syndrome
Short Stature
Short stature is a measured height of less than -2.5 standard deviations below the mean for the general population. It is a common clinical finding in patients with Turner’s syndrome (TS). In a recent study involving 176 girls with Turners syndrome, the mid-parental height was sensitive and served as a valuable tool in assessing short stature in children. Growth failure during childhood results in a predictable short stature in adult life. The average height of women with untreated TS is about 4 feet, 8 inches
Webbed neck and lymphedema
Patients with TS have a characteristic webbed neck (pterygium colli). Swelling of the hands and feet is also noted in infancy and facilitates early diagnosis of this condition.
Hypertension
Arterial hypertension has a reported prevalence of 13-58% in adult patients with TS. This confers a higher than 4-fold increase in the risk of hypertension-related mortality in this patient population.
Melanocytic nevi
Melanocytic nevi have a reported prevalence of 25-100%. These pigmented nevi tend to increase in size and number over time, with a reported increased risk of malignant transformation when they reach a size greater than 5-10mm.
Sexual infantilism
Girls with TS at the time of puberty are usually amenorrheic with no secondary sexual characteristics; however, those with mosaicism may develop primordial follicles in their ovaries, leading to estrogen-mediated breast development and hypomenorrhea
Polycystic Ovarian Syndrome
Hirsutism
Hirsutism is defined as the presence of terminal, pigmented hair in a male-pattern distribution. It is a common dermatologic manifestation of polycystic ovary syndrome (PCOS) with a reported prevalence of 50-70%
The original assessment of hirsutism using the Ferriman-Gallway score assigned a grade ranging from 0 to 4, depending on the presence and extent of terminal hair distribution involving 11 anatomic sites.
The forearm and lower leg have been excluded in the modified score since terminal hair growth in these areas is inconsistently associated with hyperandrogenemia. The modified Ferriman-Gallway score objectively assesses 9 anatomic regions for evidence of terminal hair growth. A modified Ferriman-Gallway score of ≥8 is consistent with clinically significant hirsutism.
Endocrine conditions associated with virilization
- Adrenal tumors
- Cushing’s syndrome
- Classic and non-classic congenital adrenal hyperplasia
- Hyperandrogenism, insulin resistance, acanthosis nigricans (HAIR-AN syndrome)
- Ovarian hyperthecosis
- Ovarian tumors
- The features of virilization include acne, male pattern baldness, deep male voice, and clitoral enlargement.
Acanthosis nigricans
Acanthosis nigricans (AN) is a classic dermatologic manifestation of PCOS and other endocrinopathies associated with insulin resistance. It is a velvety, dark, and plaque-like skin lesion which has a predilection for flexural areas such as the neck and axillary regions. 50% of patients with the classic obese PCOS phenotype have AN.
Acne
Acne is a common skin manifestation of PCOS, with a highly variable prevalence, based on ethnicity. Asian Indians have the highest prevalence, with the lowest being among Pacific Islanders.
Obesity
The prevalence of obesity in patients with PCOS is between 40 to 80%. In contrast to women outside the United States(US), PCOS patients in the US have relatively higher body mass indexes (BMI’s)
Male Hypogonadism
Decreased Testicular Volume
Testicular volume (TV) is a valuable surrogate marker of testosterone production and spermiogenesis function. Decreased testicular volume is associated with male hypogonadism and is more likely in primary hypogonadism compared to secondary hypogonadism.
The Prader orchidometer is an objective means of assessing testicular volume clinically. A cross-sectional study of over 400 subjects showed a strong, statistically significant positive correlation between clinical assessment of testicular volume using an orchidometer and ultrasound estimates.
Considerable tact and excellent reassurance skills are essential when using an orchidometer in order to prevent the patient from feeling inadequate.
A recent study evaluated the association between serum testosterone levels and adult testicular volumes in patients with either primary or secondary hypogonadism. A testicular volume of >30cc, estimated by an orchidometer combined with BMI measurements, had a high predictive value in assessing if testosterone levels were optimal. TV combined with BMI had a sensitivity and specificity of 85.3% and 86.5%, respectively.
Gynecomastia
Gynecomastia is the presence of palpable glandular breast tissue in men and should be differentiated from pseudogynecomastia, which is an enlargement of the breasts due to an accumulation of fat. Gynecomastia is a reported finding in male hypogonadism.
Loss of height or fragility fractures
Male hypogonadism contributes to a low bone mineral density, which can present as a fragility fracture. The prevalence of secondary osteoporosis or osteopenia in patients with male hypogonadism remains unclear at this time.
Change in body composition
Patients with male hypogonadism are predisposed to low muscle mass and strength is mainly in the lower extremities.
For the most part, hypogonadism is associated with an increase in abdominal fat, which explains the increased waist to hip ratio seen in subjects with hypogonadism.
Estrogen Resistance
Tall stature
Tall stature was described in an index case of estrogen resistance in a male patient. A recent case series including two females and one male, however, reported tall stature as an inconsistent finding in estrogen resistance.
Estrogen resistance is a rare condition with very few published case reports, making the exact estimation of prevalence uncertain.
Acanthosis nigricans
Acanthosis nigricans (AN) was reported in an index case estrogen resistance in a male patient. The lesions were distributed in the bilateral axillae.
Complete Androgen Insensitivity
Abnormalities of the external and internal genitalia
Patients with complete androgen insensitivity have a short vagina with an absent cervix on pelvic examination. Due to the normal-appearing female external genitalia, the diagnosis is often delayed.
Normal breast tissue development
Patients with CAIS usually have normal female breast tissue development. Aberrant breast tissue anywhere along the mammary line have been reported as well. There is spontaneous breast development around puberty; as such, patients can grow up with a female gender identity.